 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Dora Angyal | + 5374 word(s) | 5374 | 2021-12-28 06:46:46 | | | |
| 2 | Camila Xu | Meta information modification | 5374 | 2022-01-17 02:51:12 | | |
Video Upload Options
CFTR, the cystic fibrosis (CF) gene-encoded epithelial anion channel, has a prominent role in driving chloride, bicarbonate and fluid secretion in the ductal cells of the exocrine pancreas. Here, we summarize recent insights into the mechanism and regulation of CFTR-mediated and modulated bicarbonate secretion in the pancreatic duct, including the role of the osmotic stress/chloride sensor WNK1 and the scaffolding protein IRBIT, and current knowledge about the role of CFTR in genetic and acquired forms of pancreatitis. Furthermore, we discuss the perspectives for CFTR modulator therapy in the treatment of exocrine pancreatic insufficiency and pancreatitis and introduce pancreatic organoids as a promising model system to study CFTR function in the human pancreas, its role in the pathology of pancreatitis and its sensitivity to CFTR modulators on a personalized basis.
1. Introduction
2. Bicarbonate Transport in the Exocrine Pancreas
2.1. CFTR Is Indispensable for the Accumulation of Bicarbonate in Pancreatic Juice
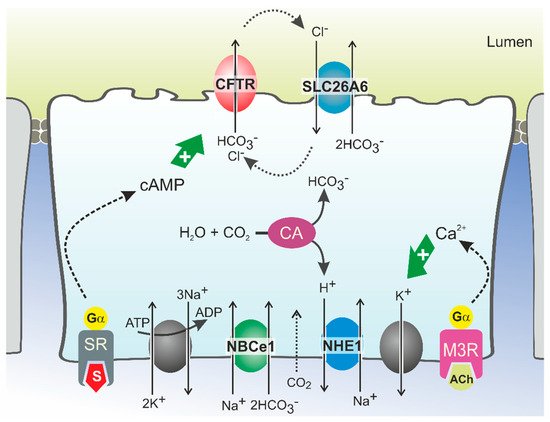
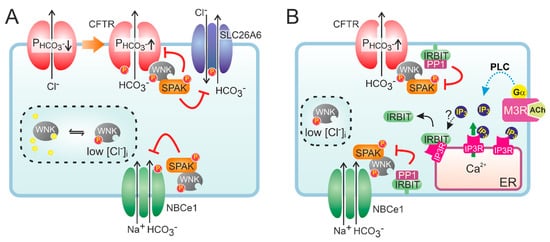
2.2. Molecular Mechanisms of CFTR-Dependent Bicarbonate Transport
3. Bicarbonate Transport in Subjects with CFTR Mutations
3.1. Impaired Pancreatic Bicarbonate Transport in CF Patients
3.2. Bicarbonate and Viscid Mucus
3.3. Bicarbonate Transport in Subjects with Non-CF-Causing CFTR Mutations
4. Acquired CFTR Dysfunction in Pancreatitis
4.1. Alcohol
4.2. Bile Acids
4.3. Smoking
4.4. Susceptibility to Pancreatitis Inducers
5. CFTR Modulators
5.1. Modulator Effects in the Pancreas
5.2. Modulator Effects on HCO3− Transport
5.3. Modulator Studies in Pancreatic Ductal Organoids

6. Concluding Remarks
References
- Shteinberg, M.; Haq, I.J.; Polineni, D.; Davies, J.C. Cystic fibrosis. Lancet 2021, 397, 2195–2211.
- Burgel, P.R.; Bellis, G.; Olesen, H.V.; Viviani, L.; Zolin, A.; Blasi, F.; Elborn, J.S. Future trends in cystic fibrosis demography in 34 European countries. Eur. Respir. J. 2015, 46, 133–141.
- Andersen, D.H. Cystic fibrosis of the pancreas and its relation to celiac disease: A clinical and pathologic study. Am. J. Dis. Child. 1938, 56, 344–399.
- Singh, V.K.; Schwarzenberg, S.J. Pancreatic insufficiency in cystic fibrosis. J. Cyst. Fibros. 2017, 16, S70–S78.
- Mayerle, J.; Sendler, M.; Hegyi, E.; Beyer, G.; Lerch, M.M.; Sahin-Tóth, M. Genetics, cell biology, and pathophysiology of pancreatitis. Gastroenterology 2019, 156, 1951–1968.e1951.
- Peery, A.F.; Crockett, S.D.; Murphy, C.C.; Lund, J.L.; Dellon, E.S.; Williams, J.L.; Jensen, E.T.; Shaheen, N.J.; Barritt, A.S.; Lieber, S.R.; et al. Burden and cost of gastrointestinal, liver, and pancreatic diseases in the United States: Update 2018. Gastroenterology 2019, 156, 254–272.e211.
- Petrov, M.S.; Yadav, D. Global epidemiology and holistic prevention of pancreatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 175–184.
- Whitcomb, D. Framework for interpretation of genetic variations in pancreatitis patients. Front. Physiol. 2012, 3, 440.
- Whitcomb, D.C.; North American Pancreatitis Study Group. Pancreatitis: TIGAR-O version 2 risk/etiology checklist with topic reviews, updates, and use primers. Clin. Transl. Gastroenterol. 2019, 10, e00027.
- Balázs, A.; Hegyi, P. Cystic fibrosis-style changes in the early phase of pancreatitis. Clin. Res. Hepatol. Gastroenterol. 2015, 39, S12–S17.
- Ooi, C.Y.; Dorfman, R.; Cipolli, M.; Gonska, T.; Castellani, C.; Keenan, K.; Freedman, S.D.; Zielenski, J.; Berthiaume, Y.; Corey, M.; et al. Type of CFTR mutation determines risk of pancreatitis in patients with cystic fibrosis. Gastroenterology 2011, 140, 153–161.
- Miller, A.C.; Comellas, A.P.; Hornick, D.B.; Stoltz, D.A.; Cavanaugh, J.E.; Gerke, A.K.; Welsh, M.J.; Zabner, J.; Polgreen, P.M. Cystic fibrosis carriers are at increased risk for a wide range of cystic fibrosis-related conditions. Proc. Natl. Acad. Sci. USA 2020, 117, 1621–1627.
- Lee, M.G.; Ohana, E.; Park, H.W.; Yang, D.; Muallem, S. Molecular mechanism of pancreatic and salivary gland fluid and HCO3-secretion. Physiol. Rev. 2012, 92, 39–74.
- Quinton, P.M. The neglected ion: HCO3-. Nat. Med. 2001, 7, 292–293.
- Hug, M.J.; Clarke, L.L.; Gray, M.A. How to measure CFTR-dependent bicarbonate transport: From single channels to the intact epithelium. Methods Mol. Biol. 2011, 741, 489–509.
- Ishiguro, H.; Yamamoto, A.; Nakakuki, M.; Yi, L.; Ishiguro, M.; Yamaguchi, M.; Kondo, S.; Mochimaru, Y. Physiology and pathophysiology of bicarbonate secretion by pancreatic duct epithelium. Nagoya J. Med. Sci. 2012, 74, 1–18.
- Petersen, O.H. Physiology of acinar cell secretion. In The Pancreas: An Integrated Textbook of Basic Science, Medicine and Surgery, 3rd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2018; pp. 41–55.
- Gullo, L.; Priori, P.; Pezzilli, R.; Biliotti, G.; Mattioli, G.; Barbara, L. Pancreatic secretory response to ordinary meals: Studies with pure pancreatic juice. Gastroenterology 1988, 94, 428–433.
- Bernacki, E.; Podkowicz, K.; Rublewska, M.; Klepacki, A. Studies on the exocrine function of healthy human pancreas: Pancreatic juice and its certain components. Acta Physiol. Pol. 1976, 27, 337–345.
- Lee, M.G.; Wigley, W.C.; Zeng, W.; Noel, L.E.; Marino, C.R.; Thomast, P.J.; Muallem, S. Regulation of Cl-/ HCO3- exchange by cystic fibrosis transmembrane conductance regulator expressed in NIH 3T3 and HEK 293 cells. J. Biol. Chem. 1999, 274, 3414–3421.
- Domschke, S.; Domschke, W.; Rösch, W.; Konturek, S.J.; Wünsch, E.; Demling, L. Bicarbonate and cyclic AMP content of pure human pancreatic juice in response to graded doses of synthetic secretin. Gastroenterology 1976, 70, 533–536.
- Sewell, W.A.; Young, J.A. Secretion of electrolytes by the pancreas of the anaestetized rat. J. Physiol. 1975, 252, 379–396.
- Mangos, J.A.; McSherry, N.R.; Nousia-Arvanitakis, S.; Irwin, K. Secretion and transductal fluxes of ions in exocrine glands of the mouse. Am. J. Physiol. 1973, 225, 18–24.
- Steward, M.C.; Ishiguro, H.; Case, R.M. Mechanisms of bicarbonate secretion in the pancreatic duct. Annu. Rev. Physiol. 2005, 67, 377–409.
- Padfield, P.J.; Garner, A.; Case, R.M. Patterns of pancreatic secretion in the anaesthetised guinea pig following stimulation with secretin, cholecystokinin octapeptide, or bombesin. Pancreas 1989, 4, 204–209.
- Stewart, A.K.; Shmukler, B.E.; Vandorpe, D.H.; Reimold, F.; Heneghan, J.F.; Nakakuki, M.; Akhavein, A.; Ko, S.; Ishiguro, H.; Alper, S.L. SLC26 anion exchangers of guinea pig pancreatic duct: Molecular cloning and functional characterization. Am. J. Physiol. Cell Physiol. 2011, 301, C289–C303.
- Yamaguchi, M.; Steward, M.C.; Smallbone, K.; Sohma, Y.; Yamamoto, A.; Ko, S.B.H.; Kondo, T.; Ishiguro, H. Bicarbonate-rich fluid secretion predicted by a computational model of guinea-pig pancreatic duct epithelium. J. Physiol. 2017, 595, 1947–1972.
- Quinton, P.M. Too much salt, too little soda: Cystic fibrosis. Sheng Li Xue Bao 2007, 59, 397–415.
- Fong, P. CFTR–SLC26 transporter interactions in epithelia. Biophys. Rev. 2012, 4, 107–116.
- Lohi, H.; Lamprecht, G.; Markovich, D.; Heil, A.; Kujala, M.; Seidler, U.; Kere, J. Isoforms of SLC26A6 mediate anion transport and have functional PDZ interaction domains. Am. J. Physiol. Cell. Physiol. 2003, 284, C769–C779.
- Novak, I.; Greger, R. Properties of the luminal membrane of isolated perfused rat pancreatic ducts. Pflugers Arch. 1988, 411, 546–553.
- Wine, J.J. Cystic fibrosis: The ‘bicarbonate before chloride’ hypothesis. Curr. Biol. 2001, 11, R463–R466.
- Namkung, W.; Lee, J.A.; Ahn, W.; Han, W.; Kwon, S.W.; Ahn, D.S.; Kim, K.H.; Lee, M.G. Ca2+ activates cystic fibrosis transmembrane conductance regulator- and Cl--dependent HCO3- transport in pancreatic duct cells. J. Biol. Chem. 2003, 278, 200–207.
- Ko, S.B.H.; Zeng, W.; Dorwart, M.R.; Luo, X.; Kim, K.H.; Millen, L.; Goto, H.; Naruse, S.; Soyombo, A.; Thomas, P.J. Gating of CFTR by the STAS domain of SLC26 transporters. Nat. Cell Biol. 2004, 6, 343–350.
- Berg, P.; Svendsen, S.L.; Sorensen, M.V.; Larsen, C.K.; Andersen, J.F.; Jensen-Fangel, S.; Jeppesen, M.; Schreiber, R.; Cabrita, I.; Kunzelmann, K.; et al. Impaired renal HCO3- excretion in cystic fibrosis. J. Am. Soc. Nephrol. 2020, 31, 1711–1727.
- Sohma, Y.; Gray, M.A.; Imai, Y.; Argent, B.E. HCO3- transport in a mathematical model of the pancreatic ductal epithelium. J. Membr. Biol. 2000, 176, 77–100.
- Illek, B.; Yankaskas, J.R.; Machen, T.E. cAMP and genistein stimulate HCO3-conductance through CFTR in human airway epithelia. Am. J. Physiol. 1997, 272, L752–L761.
- Poulsen, J.H.; Fischer, H.; Illek, B.; Machen, T.E. Bicarbonate conductance and pH regulatory capability of cystic fibrosis transmembrane conductance regulator. Proc. Natl. Acad. Sci. USA 1994, 91, 5340–5344.
- Linsdell, P.; Tabcharani, J.A.; Rommens, J.M.; Hou, Y.-X.; Chang, X.-B.; Tsui, L.-C.; Riordan, J.R.; Hanrahan, J.W. Permeability of wild-type and mutant cystic fibrosis transmembrane conductance regulator chloride channels to polyatomic anions. J. Gen. Physiol. 1997, 110, 355–364.
- Ishiguro, H.; Steward, M.C.; Naruse, S.; Ko, S.B.; Goto, H.; Case, R.M.; Kondo, T.; Yamamoto, A. CFTR functions as a bicarbonate channel in pancreatic duct cells. J. Gen. Physiol. 2009, 133, 315–326.
- Ferrera, L.; Baroni, D.; Moran, O. Lumacaftor-rescued F508del-CFTR has a modified bicarbonate permeability. J. Cyst. Fibros. 2019, 18, 602–605.
- Tang, L.; Fatehi, M.; Linsdell, P. Mechanism of direct bicarbonate transport by the CFTR anion channel. J. Cyst. Fibros. 2009, 8, 115–121.
- Reddy, M.M.; Quinton, P.M. Selective activation of cystic fibrosis transmembrane conductance regulator Cl- and HCO3- conductances. J. Pancreas 2001, 2, 212–218.
- Reddy, M.M.; Quinton, P.M. Control of dynamic CFTR selectivity by glutamate and ATP in epithelial cells. Nature 2003, 423, 756–760.
- Shcheynikov, N.; Kim, K.H.; Kim, K.M.; Dorwart, M.R.; Ko, S.B.H.; Goto, H.; Naruse, S.; Thomas, P.J.; Muallem, S. Dynamic control of cystic fibrosis transmembrane conductance regulator Cl-/HCO3- selectivity by external Cl-. J Biol. Chem. 2004, 279, 21857–21865.
- Wright, A.M.; Gong, X.; Verdon, B.; Linsdell, P.; Mehta, A.; Riordan, J.R.; Argent, B.E.; Gray, M.A. Novel regulation of cystic fibrosis transmembrane conductance regulator (CFTR) channel gating by external chloride. J. Biol. Chem. 2004, 279, 41658–41663.
- Pallagi, P.; Hegyi, P.; Rakonczay, Z. The physiology and pathophysiology of pancreatic ductal secretion: The background for clinicians. Pancreas 2015, 44, 1211–1233.
- Park, H.W.; Lee, M.G. Transepithelial bicarbonate secretion: Lessons from the pancreas. Cold Spring Harb. Perspect. Med. 2012, 2, a009571.
- Delpire, E.; Gagnon, K.B.E. SPAK and OSR1: STE20 kinases involved in the regulation of ion homoeostasis and volume control in mammalian cells. Biochem. J. 2008, 409, 321–331.
- Park, H.W.; Nam, J.H.; Kim, J.Y.; Namkung, W.; Yoon, J.S.; Lee, J.S.; Kim, K.S.; Venglovecz, V.; Gray, M.A.; Kim, K.H.; et al. Dynamic regulation of CFTR bicarbonate permeability by i and its role in pancreatic bicarbonate secretion. Gastroenterology 2010, 139, 620–631.
- Kim, Y.; Jun, I.; Shin, D.H.; Yoon, J.G.; Piao, H.; Jung, J.; Park, H.W.; Cheng, M.H.; Bahar, I.; Whitcomb, D.C.; et al. Regulation of CFTR bicarbonate channel activity by WNK1: Implications for pancreatitis and CFTR-related disorders. Cell. Mol. Gastroenterol. Hepatol 2020, 9, 79–103.
- Dorwart, M.R.; Shcheynikov, N.; Wang, Y.; Stippec, S.; Muallem, S. SLC26A9 is a Cl− channel regulated by the WNK kinases. J. Physiol. 2007, 584, 333–345.
- Yang, C.-L.; Liu, X.; Paliege, A.; Zhu, X.; Bachmann, S.; Dawson, D.C.; Ellison, D.H. WNK1 and WNK4 modulate CFTR activity. Biochem. Biophys. Res. Commun. 2007, 353, 535–540.
- Yang, D.; Li, Q.; So, I.; Huang, C.L.; Ando, H.; Mizutani, A.; Seki, G.; Mikoshiba, K.; Thomas, P.J.; Muallem, S. IRBIT governs epithelial secretion in mice by antagonizing the WNK/SPAK kinase pathway. J. Clin. Investig. 2011, 121, 956–965.
- Hong, J.H.; Yang, D.; Shcheynikov, N.; Ohana, E.; Shin, D.M.; Muallem, S. Convergence of IRBIT, phosphatidylinositol (4,5) bisphosphate, and WNK/SPAK kinases in regulation of the Na+-HCO3- cotransporters family. Proc. Natl. Acad. Sci. USA 2013, 110, 4105–4110.
- Shirakabe, K.; Priori, G.; Yamada, H.; Ando, H.; Horita, S.; Fujita, T.; Fujimoto, I.; Mizutani, A.; Seki, G.; Mikoshiba, K. IRBIT, an inositol 1,4,5-trisphosphate receptor-binding protein, specifically binds to and activates pancreas-type Na+/HCO3- cotransporter 1 (pNBC1). Proc. Natl. Acad. Sci. USA 2006, 103, 9542–9547.
- Yang, D.; Shcheynikov, N.; Zeng, W.; Ohana, E.; So, I.; Ando, H.; Mizutani, A.; Mikoshiba, K.; Muallem, S. IRBIT coordinates epithelial fluid and HCO3- secretion by stimulating the transporters pNBC1 and CFTR in the murine pancreatic duct. J. Clin. Investig. 2009, 119, 193–202.
- Hong, J.H.; Park, S.; Shcheynikov, N.; Muallem, S. Mechanism and synergism in epithelial fluid and electrolyte secretion. Pflugers Arch. 2014, 466, 1487–1499.
- Park, S.; Shcheynikov, N.; Hong, J.H.; Zheng, C.; Suh, S.H.; Kawaai, K.; Ando, H.; Mizutani, A.; Abe, T.; Kiyonari, H.; et al. Irbit mediates synergy between ca(2+) and cAMP signaling pathways during epithelial transport in mice. Gastroenterology 2013, 145, 232–241.
- Rosendahl, J.; Witt, H.; Szmola, R.; Bhatia, E.; Ozsvári, B.; Landt, O.; Schulz, H.U.; Gress, T.M.; Pfützer, R.; Löhr, M.; et al. Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat. Genet. 2008, 40, 78–82.
- Rosendahl, J.; Landt, O.; Bernadova, J.; Kovacs, P.; Teich, N.; Bödeker, H.; Keim, V.; Ruffert, C.; Mössner, J.; Kage, A.; et al. CFTR, SPINK1, CTRC and PRSS1 variants in chronic pancreatitis: Is the role of mutated CFTR overestimated? Gut 2013, 62, 582–592.
- Rosendahl, J.; Kirsten, H.; Hegyi, E.; Kovacs, P.; Weiss, F.U.; Laumen, H.; Lichtner, P.; Ruffert, C.; Chen, J.M.; Masson, E.; et al. Genome-wide association study identifies inversion in the CTRB1-CTRB2 locus to modify risk for alcoholic and non-alcoholic chronic pancreatitis. Gut 2018, 67, 1855–1863.
- Witt, H.; Beer, S.; Rosendahl, J.; Chen, J.M.; Chandak, G.R.; Masamune, A.; Bence, M.; Szmola, R.; Oracz, G.; Macek, M., Jr.; et al. Variants in CPA1 are strongly associated with early onset chronic pancreatitis. Nat. Genet. 2013, 45, 1216–1220.
- Fjeld, K.; Weiss, F.U.; Lasher, D.; Rosendahl, J.; Chen, J.M.; Johansson, B.B.; Kirsten, H.; Ruffert, C.; Masson, E.; Steine, S.J.; et al. A recombined allele of the lipase gene CEL and its pseudogene CELP confers susceptibility to chronic pancreatitis. Nat. Genet. 2015, 47, 518–522.
- Cavestro, G.M.; Zuppardo, R.A.; Bertolini, S.; Sereni, G.; Frulloni, L.; Okolicsanyi, S.; Calzolari, C.; Singh, S.K.; Sianesi, M.; Del Rio, P.; et al. Connections between genetics and clinical data: Role of MCP-1, CFTR, and SPINK-1 in the setting of acute, acute recurrent, and chronic pancreatitis. Am. J. Gastroenterol. 2010, 105, 199–206.
- Sultan, M.; Werlin, S.; Venkatasubramani, N. Genetic prevalence and characteristics in children with recurrent pancreatitis. J. Pediatr. Gastroenterol. Nutr. 2012, 54, 645–650.
- Werlin, S.; Konikoff, F.M.; Halpern, Z.; Barkay, O.; Yerushalmi, B.; Broide, E.; Santo, E.; Shamir, R.; Shaoul, R.; Shteyer, E.; et al. Genetic and electrophysiological characteristics of recurrent acute pancreatitis. J. Pediatr. Gastroenterol. Nutr. 2015, 60, 675–679.
- Sharer, N.; Schwarz, M.; Malone, G.; Howarth, A.; Painter, J.; Super, M.; Braganza, J. Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. N. Engl. J. Med. 1998, 339, 645–652.
- Cohn, J.A.; Noone, P.G.; Jowell, P.S. Idiopathic pancreatitis related to CFTR: Complex inheritance and identification of a modifier gene. J. Investig. Med. 2002, 50, 247S–255S.
- Weiss, F.U.; Simon, P.; Bogdanova, N.; Mayerle, J.; Dworniczak, B.; Horst, J.; Lerch, M.M. Complete cystic fibrosis transmembrane conductance regulator gene sequencing in patients with idiopathic chronic pancreatitis and controls. Gut 2005, 54, 1456–1460.
- Cohn, J.A.; Friedman, K.J.; Noone, P.G.; Knowles, M.R.; Silverman, L.M.; Jowell, P.S. Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis. N. Engl. J. Med. 1998, 339, 653–658.
- Kumar, S.; Ooi, C.Y.; Werlin, S.; Abu-El-Haija, M.; Barth, B.; Bellin, M.D.; Durie, P.R.; Fishman, D.S.; Freedman, S.D.; Gariepy, C.; et al. Risk factors associated with pediatric acute recurrent and chronic pancreatitis: Lessons from INSPPIRE. JAMA Pediatr. 2016, 170, 562–569.
- Beyer, G.; Habtezion, A.; Werner, J.; Lerch, M.M.; Mayerle, J. Chronic pancreatitis. Lancet 2020, 396, 499–512.
- Weiss, F.U.; Skube, M.E.; Lerch, M.M. Chronic pancreatitis: An update on genetic risk factors. Curr. Opin. Gastroenterol. 2018, 34, 322–329.
- Gastard, J.; Joubaud, F.; Farbos, T.; Loussouarn, J.; Marion, J.; Pannier, M.; Renaudet, F.; Valdazo, R.; Gosselin, M. Etiology and course of primary chronic pancreatitis in western France. Digestion 1973, 9, 416–428.
- Yadav, D.; Lowenfels, A.B. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology 2013, 144, 1252–1261.
- Coté, G.A.; Yadav, D.; Slivka, A.; Hawes, R.H.; Anderson, M.A.; Burton, F.R.; Brand, R.E.; Banks, P.A.; Lewis, M.D.; Disario, J.A. Alcohol and smoking as risk factors in an epidemiology study of patients with chronic pancreatitis. Clin. Gastroenterol. Hepatol. 2011, 9, 266–273.
- Yadav, D.; Hawes, R.H.; Brand, R.E.; Anderson, M.A.; Money, M.E.; Banks, P.A.; Bishop, M.D.; Baillie, J.; Sherman, S.; DiSario, J.; et al. Alcohol consumption, cigarette smoking, and the risk of recurrent acute and chronic pancreatitis. Arch. Intern. Med. 2009, 169, 1035–1045.
- Frulloni, L.; Gabbrielli, A.; Pezzilli, R.; Zerbi, A.; Cavestro, G.M.; Marotta, F.; Falconi, M.; Gaia, E.; Uomo, G.; Maringhini, A.; et al. Chronic pancreatitis: Report from a multicenter Italian survey (PanCroInfAISP) on 893 patients. Dig. Liver Dis. 2009, 41, 311–317.
- Lankisch, P.G.; Assmus, C.; Maisonneuve, P.; Lowenfels, A.B. Epidemiology of pancreatic diseases in Lüneburg County: A study in a defined German population. Pancreatology 2002, 2, 469–477.
- Munigala, S.; Conwell, D.L.; Gelrud, A.; Agarwal, B. Heavy smoking is associated with lower age at first episode of acute pancreatitis and a higher risk of recurrence. Pancreas 2015, 44, 876–881.
- Ren, C.L.; Borowitz, D.S.; Gonska, T.; Howenstine, M.S.; Levy, H.; Massie, J.; Milla, C.; Munck, A.; Southern, K.W. Cystic fibrosis transmembrane conductance regulator-related metabolic syndrome and cystic fibrosis screen positive, inconclusive diagnosis. J. Pediatr. 2017, 181, S45–S51.e41.
- Munck, A.; Mayell, S.J.; Winters, V.; Shawcross, A.; Derichs, N.; Parad, R.; Barben, J.; Southern, K.W. Cystic fibrosis screen positive, inconclusive diagnosis (CFSPID): A new designation and management recommendations for infants with an inconclusive diagnosis following newborn screening. J. Cyst. Fibros. 2015, 14, 706–713.
- Johansen, P.; Anderson, C.; Hadorn, B. Cystic fibrosis of the pancreas: A generalised disturbance of water and electrolyte movement in exocrine tissues. Lancet 1968, 291, 455–460.
- Kopelman, H.; Forstner, G.; Durie, P.; Corey, M. Origins of chloride and bicarbonate secretory defects in the cystic fibrosis pancreas, as suggested by pancreatic function studies on control and CF subjects with preserved pancreatic function. Clin. Investig. Med. 1989, 12, 207–211.
- Kopelman, H.; Corey, M.; Gaskin, K.; Durie, P.; Weizman, Z.; Forstner, G. Impaired chloride secretion, as well as bicarbonate secretion, underlies the fluid secretory defect in the cystic fibrosis pancreas. Gastroenterology 1988, 95, 349–355.
- Ahmed, N.; Corey, M.; Forstner, G.; Zielenski, J.; Tsui, L.C.; Ellis, L.; Tullis, E.; Durie, P. Molecular consequences of cystic fibrosis transmembrane regulator (CFTR) gene mutations in the exocrine pancreas. Gut 2003, 52, 1159–1164.
- Taylor, C.J.; Aswani, N. The pancreas in cystic fibrosis. Paediatr. Respir. Rev. 2002, 3, 77–81.
- Choi, J.Y.; Muallem, D.; Kiselyov, K.; Lee, M.G.; Thomas, P.J.; Muallem, S. Aberrant CFTR-dependent HCO3- transport in mutations associated with cystic fibrosis. Nature 2001, 410, 94–97.
- Choi, J.Y.; Lee, M.G.; Ko, S.; Muallem, S. Cl-dependent HCO3- transport by cystic fibrosis transmembrane conductance regulator. J. Pancreas 2001, 2, 243–246.
- Shwachman, H.; Lebenthal, E.; Khaw, K.T. Recurrent acute pancreatitis in patients with cystic fibrosis with normal pancreatic enzymes. Pediatrics 1975, 55, 86–95.
- Morrison, C.B.; Markovetz, M.R.; Ehre, C. Mucus, mucins, and cystic fibrosis. Pediatr. Pulmonol. 2019, 54, S84–S96.
- Hansson, G.C. Mucus and mucins in diseases of the intestinal and respiratory tracts. J. Intern. Med. 2019, 285, 479–490.
- Garcia, M.A.S.; Yang, N.; Quinton, P.M. Normal mouse intestinal mucus release requires cystic fibrosis transmembrane regulator–dependent bicarbonate secretion. J. Clin. Investig. 2009, 119, 3497.
- Gustafsson, J.K.; Ermund, A.; Ambort, D.; Johansson, M.E.V.; Nilsson, H.E.; Thorell, K.; Hebert, H.; Sjövall, H.; Hansson, G.C. Bicarbonate and functional CFTR channel are required for proper mucin secretion and link cystic fibrosis with its mucus phenotype. J. Exp. Med. 2012, 209, 1263–1272.
- Chen, J.-H.; Stoltz, D.A.; Karp, P.H.; Ernst, S.E.; Pezzulo, A.A.; Moninger, T.O.; Rector, M.V.; Reznikov, L.R.; Launspach, J.L.; Chaloner, K. Loss of anion transport without increased sodium absorption characterizes newborn porcine cystic fibrosis airway epithelia. Cell 2010, 143, 911–923.
- Kim, D.; Liao, J.; Scales, N.B.; Martini, C.; Luan, X.; Abu-Arish, A.; Robert, R.; Luo, Y.; McKay, G.A.; Nguyen, D.; et al. Large pH oscillations promote host defense against human airways infection. J. Exp. Med. 2021, 218.
- Simonin, J.; Bille, E.; Crambert, G.; Noel, S.; Dreano, E.; Edwards, A.; Hatton, A.; Pranke, I.; Villeret, B.; Cottart, C.-H. Airway surface liquid acidification initiates host defense abnormalities in cystic fibrosis. Sci. Rep. 2019, 9, 1–11.
- Tucker, J.A.; Spock, A.; Spicer, S.S.; Shelburne, J.D.; Bradford, W. Inspissation of pancreatic zymogen material in cystic fibrosis. Ultrastruct. Pathol. 2003, 27, 323–335.
- Gibson-Corley, K.N.; Meyerholz, D.K.; Engelhardt, J.F. Pancreatic pathophysiology in cystic fibrosis. J. Pathol. 2016, 238, 311–320.
- Bombieri, C.; Claustres, M.; De Boeck, K.; Derichs, N.; Dodge, J.; Girodon, E.; Sermet, I.; Schwarz, M.; Tzetis, M.; Wilschanski, M.; et al. Recommendations for the classification of diseases as CFTR-related disorders. J. Cyst. Fibros. 2011, 10, S86–S102.
- Steiner, B.; Rosendahl, J.; Witt, H.; Teich, N.; Keim, V.; Schulz, H.-U.; Pfützer, R.; Lühr, M.; Gress, T.M.; Nickel, R.; et al. Common CFTR haplotypes and susceptibility to chronic pancreatitis and congenital bilateral absence of the vas deferens. Hum. Mutat. 2011, 32, 912–920.
- LaRusch, J.; Jung, J.; General, I.J.; Lewis, M.D.; Park, H.W.; Brand, R.E.; Gelrud, A.; Anderson, M.A.; Banks, P.A.; Conwell, D.; et al. Mechanisms of CFTR functional variants that impair regulated bicarbonate permeation and increase risk for pancreatitis but not for cystic fibrosis. PLoS Genet. 2014, 10, e1004376.
- Xu, W.M.; Shi, Q.X.; Chen, W.Y.; Zhou, C.X.; Ni, Y.; Rowlands, D.K.; Liu, G.Y.; Zhu, H.; Ma, Z.G.; Wang, X.F. Cystic fibrosis transmembrane conductance regulator is vital to sperm fertilizing capacity and male fertility. Proc. Natl. Acad. Sci. USA 2007, 104, 9816–9821.
- Martinez, B.; Heller, M.; Gaitch, N.; Hubert, D.; Burgel, P.R.; Levy, P.; Girodon, E.; Bienvenu, T. p.Arg75Gln, a CFTR variant involved in the risk of CFTR-related disorders? J. Hum. Genet. 2014, 59, 206–210.
- Naruse, S.; Kitagawa, M.; Ishiguro, H. Molecular understanding of chronic pancreatitis: A perspective on the future. Mol. Med. Today 1999, 5, 493–499.
- Yamamoto, A.; Ishiguro, H.; Ko, S.B.; Suzuki, A.; Wang, Y.; Hamada, H.; Mizuno, N.; Kitagawa, M.; Hayakawa, T.; Naruse, S. Ethanol induces fluid hypersecretion from guinea-pig pancreatic duct cells. J. Physiol. 2003, 551, 917–926.
- Maléth, J.; Balázs, A.; Pallagi, P.; Balla, Z.; Kui, B.; Katona, M.; Judák, L.; Németh, I.; Kemény, L.V.; Rakonczay, Z.; et al. Alcohol disrupts levels and function of the cystic fibrosis transmembrane conductance regulator to promote development of pancreatitis. Gastroenterology 2015, 148, 427–439.
- Judák, L.; Hegyi, P.; Rakonczay, Z., Jr.; Maléth, J.; Gray, M.A.; Venglovecz, V. Ethanol and its non-oxidative metabolites profoundly inhibit CFTR function in pancreatic epithelial cells which is prevented by ATP supplementation. Pflugers Arch. 2014, 466, 549–562.
- Pallagi, P.; Madácsy, T.; Varga, Á.; Maléth, J. Intracellular Ca2+ signalling in the pathogenesis of acute pancreatitis: Recent advances and translational perspectives. Int. J. Mol. Sci. 2020, 21, 4005.
- Huang, W.; Booth, D.M.; Cane, M.C.; Chvanov, M.; Javed, M.A.; Elliott, V.L.; Armstrong, J.A.; Dingsdale, H.; Cash, N.; Li, Y.; et al. Fatty acid ethyl ester synthase inhibition ameliorates ethanol-induced Ca2+-dependent mitochondrial dysfunction and acute pancreatitis. Gut 2014, 63, 1313–1324.
- Mederos, M.A.; Reber, H.A.; Girgis, M.D. Acute pancreatitis: A review. JAMA 2021, 325, 382–390.
- Muili, K.A.; Wang, D.; Orabi, A.I.; Sarwar, S.; Luo, Y.; Javed, T.A.; Eisses, J.F.; Mahmood, S.M.; Jin, S.; Singh, V.P. Bile acids induce pancreatic acinar cell injury and pancreatitis by activating calcineurin. J. Biol. Chem. 2013, 288, 570–580.
- Venglovecz, V.; Rakonczay, Z., Jr.; Ozsvári, B.; Takács, T.; Lonovics, J.; Varró, A.; Gray, M.A.; Argent, B.E.; Hegyi, P. Effects of bile acids on pancreatic ductal bicarbonate secretion in guinea pig. Gut 2008, 57, 1102–1112.
- Ignáth, I.; Hegyi, P.; Venglovecz, V.; Székely, C.A.; Carr, G.; Hasegawa, M.; Inoue, M.; Takács, T.; Argent, B.E.; Gray, M.A.; et al. CFTR expression but not Cl- transport is involved in the stimulatory effect of bile acids on apical Cl-/HCO3- exchange activity in human pancreatic duct cells. Pancreas 2009, 38, 921–929.
- Maléth, J.; Venglovecz, V.; Rázga, Z.; Tiszlavicz, L.; Rakonczay, Z., Jr.; Hegyi, P. Non-conjugated chenodeoxycholate induces severe mitochondrial damage and inhibits bicarbonate transport in pancreatic duct cells. Gut 2011, 60, 136–138.
- Raju, S.V.; Jackson, P.L.; Courville, C.A.; McNicholas, C.M.; Sloane, P.A.; Sabbatini, G.; Tidwell, S.; Tang, L.P.; Liu, B.; Fortenberry, J.A.; et al. Cigarette smoke induces systemic defects in cystic fibrosis transmembrane conductance regulator function. Am. J. Respir. Crit. Care Med. 2013, 188, 1321–1330.
- Hegyi, P.; Wilschanski, M.; Muallem, S.; Lukacs, G.L.; Sahin-Toth, M.; Uc, A.; Gray, M.A.; Rakonczay, Z., Jr.; Maleth, J. CFTR: A new horizon in the pathomechanism and treatment of pancreatitis. Rev. Physiol. Biochem. Pharmacol. 2016, 170, 37–66.
- Alexander, N.S.; Blount, A.; Zhang, S.; Skinner, D.; Hicks, S.B.; Chestnut, M.; Kebbel, F.A.; Sorscher, E.J.; Woodworth, B.A. Cystic fibrosis transmembrane conductance regulator modulation by the tobacco smoke toxin acrolein. Laryngoscope 2012, 122, 1193–1197.
- Barreto, S.G. How does cigarette smoking cause acute pancreatitis? Pancreatology 2016, 16, 157–163.
- Schnúr, A.; Premchandar, A.; Bagdany, M.; Lukacs, G.L. Phosphorylation-dependent modulation of CFTR macromolecular signalling complex activity by cigarette smoke condensate in airway epithelia. Sci. Rep. 2019, 9, 12706.
- Rasmussen, J.E.; Sheridan, J.T.; Polk, W.; Davies, C.M.; Tarran, R. Cigarette smoke-induced Ca2+ release leads to cystic fibrosis transmembrane conductance regulator (CFTR) dysfunction. J. Biol. Chem. 2014, 289, 7671–7681.
- Marklew, A.J.; Patel, W.; Moore, P.J.; Tan, C.D.; Smith, A.J.; Sassano, M.F.; Gray, M.A.; Tarran, R. Cigarette smoke exposure induces retrograde trafficking of CFTR to the endoplasmic reticulum. Sci. Rep. 2019, 9, 13655.
- Clunes, L.A.; Davies, C.M.; Coakley, R.D.; Aleksandrov, A.A.; Henderson, A.G.; Zeman, K.L.; Worthington, E.N.; Gentzsch, M.; Kreda, S.M.; Cholon, D.; et al. Cigarette smoke exposure induces CFTR internalization and insolubility, leading to airway surface liquid dehydration. FASEB J. 2012, 26, 533–545.
- Raju, S.V.; Lin, V.Y.; Liu, L.; McNicholas, C.M.; Karki, S.; Sloane, P.A.; Tang, L.; Jackson, P.L.; Wang, W.; Wilson, L. The cystic fibrosis transmembrane conductance regulator potentiator ivacaftor augments mucociliary clearance abrogating cystic fibrosis transmembrane conductance regulator inhibition by cigarette smoke. Am. J. Respir. Cell Mol. Biol. 2017, 56, 99–108.
- Kadiyala, V.; Lee, L.S.; Banks, P.A.; Suleiman, S.; Paulo, J.A.; Wang, W.; Rosenblum, J.; Sainani, N.I.; Mortele, K.; Conwell, D.L. Cigarette smoking impairs pancreatic duct cell bicarbonate secretion. J. Pancreas 2013, 14, 31.
- Pallagi, P.; Tálas, D.; Madácsy, T.; Venglovecz, V.; Tóth, E.; Darvasi, E.; Balla, Z.; Tóth, K.; Schnúr, A.; Maléth, J.; et al. Investigation of the pathomechanism of smoke-induced pancreatic damage. Pancreatology 2019, 19, S12.
- Ballengee, C.R.; Brooks, P.; Leong, T.; Geem, D.; Freeman, A.J. Effects of second-hand smoke on pancreatitis in children. Pancreas 2019, 48, 706–710.
- Trapp, S.; Aghdassi, A.A.; Glaubitz, J.; Sendler, M.; Weiss, F.U.; Kühn, J.P.; Kromrey, M.L.; Mahajan, U.M.; Pallagi, P.; Rakonczay, Z.; et al. Pancreatitis severity in mice with impaired CFTR function but pancreatic sufficiency is mediated via ductal and inflammatory cells-not acinar cells. J. Cell. Mol. Med. 2021, 25, 4658–4670.
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445-tezacaftor-ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N. Engl. J. Med. 2018, 379, 1612–1620.
- Capurro, V.; Tomati, V.; Sondo, E.; Renda, M.; Borrelli, A.; Pastorino, C.; Guidone, D.; Venturini, A.; Giraudo, A.; Mandrup Bertozzi, S.; et al. Partial rescue of F508del-CFTR stability and trafficking defects by double corrector treatment. Int. J. Mol. Sci. 2021, 22, 5262.
- Veit, G.; Roldan, A.; Hancock, M.A.; Da Fonte, D.F.; Xu, H.; Hussein, M.; Frenkiel, S.; Matouk, E.; Velkov, T.; Lukacs, G.L. Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (trikafta) combination. JCI Insight 2020, 5, e139983.
- Giuliano, K.A.; Wachi, S.; Drew, L.; Dukovski, D.; Green, O.; Bastos, C.; Cullen, M.D.; Hauck, S.; Tait, B.D.; Munoz, B.; et al. Use of a high-throughput phenotypic screening strategy to identify amplifiers, a novel pharmacological class of small molecules that exhibit functional synergy with potentiators and correctors. SLAS Discov. 2018, 23, 111–121.
- Prokhorova, I.; Altman, R.B.; Djumagulov, M.; Shrestha, J.P.; Urzhumtsev, A.; Ferguson, A.; Chang, C.-W.T.; Yusupov, M.; Blanchard, S.C.; Yusupova, G. Aminoglycoside interactions and impacts on the eukaryotic ribosome. Proc. Natl. Acad. Sci. USA 2017, 114, E10899–E10908.
- Kerem, E. ELX-02: An investigational read-through agent for the treatment of nonsense mutation-related genetic disease. Expert Opin. Investig. Drugs 2020, 29, 1347–1354.
- de Poel, E.; Spelier, S.; Suen, S.W.F.; Kruisselbrink, E.; Graeber, S.Y.; Mall, M.A.; Weersink, E.J.M.; van der Eerden, M.M.; Koppelman, G.H.; van der Ent, C.K.; et al. Functional restoration of CFTR nonsense mutations in intestinal organoids. J. Cyst. Fibros. 2021, in press.
- Sharma, J.; Du, M.; Wong, E.; Mutyam, V.; Li, Y.; Chen, J.; Wangen, J.; Thrasher, K.; Fu, L.; Peng, N.; et al. A small molecule that induces translational readthrough of CFTR nonsense mutations by eRF1 depletion. Nat. Commun. 2021, 12, 4358.
- Bardin, E.; Pastor, A.; Semeraro, M.; Golec, A.; Hayes, K.; Chevalier, B.; Berhal, F.; Prestat, G.; Hinzpeter, A.; Gravier-Pelletier, C.; et al. Modulators of CFTR. Updates on clinical development and future directions. Eur. J. Med. Chem. 2021, 213, 113195.
- Fűr, G.; Bálint, E.R.; Orján, E.M.; Balla, Z.; Kormányos, E.S.; Czira, B.; Szűcs, A.; Kovács, D.P.; Pallagi, P.; Maléth, J.; et al. Mislocalization of CFTR expression in acute pancreatitis and the beneficial effect of VX-661 + VX-770 treatment on disease severity. J. Physiol. 2021, 599, 4955–4971.
- Zeng, M.; Szymczak, M.; Ahuja, M.; Zheng, C.; Yin, H.; Swaim, W.; Chiorini, J.A.; Bridges, R.J.; Muallem, S. Restoration of CFTR activity in ducts rescues acinar cell function and reduces inflammation in pancreatic and salivary glands of mice. Gastroenterology 2017, 153, 1148–1159.
- Bose, S.J.; Bijvelds, M.J.C.; Wang, Y.; Liu, J.; Cai, Z.; Bot, A.G.M.; de Jonge, H.R.; Sheppard, D.N. Differential thermostability and response to cystic fibrosis transmembrane conductance regulator potentiators of human and mouse F508del-CFTR. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L71–L86.
- Sellers, Z.M. Pancreatic complications in children with cystic fibrosis. Curr. Opin. Pediatr. 2020, 32, 661–667.
- Gariepy, C.E.; Ooi, C.Y.; Maqbool, A.; Ellery, K.M. Demographics and risk factors for pediatric recurrent acute pancreatitis. Curr. Opin. Gastroenterol. 2021, 37.
- Hoppe, J.E.; Chilvers, M.; Ratjen, F.; McNamara, J.J.; Owen, C.A.; Tian, S.; Zahigian, R.; Cornell, A.G.; McColley, S.A. Long-term safety of lumacaftor–ivacaftor in children aged 2–5 years with cystic fibrosis homozygous for the F508del-CFTR mutation: A multicentre, phase 3, open-label, extension study. Lancet Respir. Med. 2021, 9, 977–988.
- Akshintala, V.S.; Kamal, A.; Faghih, M.; Cutting, G.R.; Cebotaru, L.; West, N.E.; Jennings, M.T.; Dezube, R.; Whitcomb, D.C.; Lechtzin, N.; et al. Cystic fibrosis transmembrane conductance regulator modulators reduce the risk of recurrent acute pancreatitis among adult patients with pancreas sufficient cystic fibrosis. Pancreatology 2019, 19, 1023–1026.
- Carrion, A.; Borowitz, D.S.; Freedman, S.D.; Siracusa, C.M.; Goralski, J.L.; Hadjiliadis, D.; Srinivasan, S.; Stokes, D.C. Reduction of recurrence risk of pancreatitis in cystic fibrosis with ivacaftor: Case series. J. Pediatr. Gastroenterol. Nutr. 2018, 66, 451–454.
- Ramsey, M.L.; Gokun, Y.; Sobotka, L.A.; Wellner, M.R.; Porter, K.; Kirkby, S.E.; Li, S.S.; Papachristou, G.I.; Krishna, S.G.; Stanich, P.P.; et al. Cystic fibrosis transmembrane conductance regulator modulator use is associated with reduced pancreatitis hospitalizations in patients with cystic fibrosis. Am. J. Gastroenterol. 2021, in press.
- Johns, J.D.; Rowe, S.M. The effect of CFTR modulators on a cystic fibrosis patient presenting with recurrent pancreatitis in the absence of respiratory symptoms: A case report. BMC Gastroenterol. 2019, 19, 1–4.
- Gelfond, D.; Heltshe, S.; Ma, C.; Rowe, S.M.; Frederick, C.; Uluer, A.; Sicilian, L.; Konstan, M.; Tullis, E.; Roach, R.N.C. Impact of CFTR modulation on intestinal pH, motility, and clinical outcomes in patients with cystic fibrosis and the G551D mutation. Clin. Transl. Gastroenterol. 2017, 8, e81.
- Rehman, T.; Karp, P.H.; Tan, P.; Goodell, B.J.; Pezzulo, A.A.; Thurman, A.L.; Thornell, I.M.; Durfey, S.L.; Duffey, M.E.; Stoltz, D.A.; et al. Inflammatory cytokines TNF-α and IL-17 enhance the efficacy of cystic fibrosis transmembrane conductance regulator modulators. J. Clin. Investig. 2021, 131, e150398.
- Fiore, M.; Picco, C.; Moran, O. Correctors modify the bicarbonate permeability of F508del-CFTR. Sci. Rep. 2020, 10, 8440.
- Laselva, O.; Moraes, T.J.; He, G.; Bartlett, C.; Szàrics, I.; Ouyang, H.; Gunawardena, T.N.A.; Strug, L.; Bear, C.E.; Gonska, T. The CFTR mutation c.3453G>C (D1152H) confers an anion selectivity defect in primary airway tissue that can be rescued by ivacaftor. J. Pers. Med. 2020, 10, 40.
- Dekkers, J.F.; Berkers, G.; Kruisselbrink, E.; Vonk, A.; de Jonge, H.R.; Janssens, H.M.; Bronsveld, I.; van de Graaf, E.A.; Nieuwenhuis, E.E.; Houwen, R.H.; et al. Characterizing responses to CFTR-modulating drugs using rectal organoids derived from subjects with cystic fibrosis. Sci. Transl. Med. 2016, 8, 344ra384.
- de Winter-de Groot, K.M.; Janssens, H.M.; van Uum, R.T.; Dekkers, J.F.; Berkers, G.; Vonk, A.; Kruisselbrink, E.; Oppelaar, H.; Vries, R.; Clevers, H.; et al. Stratifying infants with cystic fibrosis for disease severity using intestinal organoid swelling as a biomarker of CFTR function. Eur. Respir. J. 2018, 52, 1702529.
- Berkers, G.; van Mourik, P.; Vonk, A.M.; Kruisselbrink, E.; Dekkers, J.F.; de Winter-de Groot, K.M.; Arets, H.G.M.; Marck-van der Wilt, R.E.P.; Dijkema, J.S.; Vanderschuren, M.M.; et al. Rectal organoids enable personalized treatment of cystic fibrosis. Cell Rep. 2019, 26, 1701–1708 e1703.
- Georgakopoulos, N.; Prior, N.; Angres, B.; Mastrogiovanni, G.; Cagan, A.; Harrison, D.; Hindley, C.J.; Arnes-Benito, R.; Liau, S.-S.; Curd, A. Long-term expansion, genomic stability and in vivo safety of adult human pancreas organoids. BMC Dev. Biol. 2020, 20, 1–20.
- Driehuis, E.; Gracanin, A.; Vries, R.G.J.; Clevers, H.; Boj, S.F. Establishment of pancreatic organoids from normal tissue and tumors. STAR Protoc. 2020, 1, 100192.
- Shik Mun, K.; Arora, K.; Huang, Y.; Yang, F.; Yarlagadda, S.; Ramananda, Y.; Abu-El-Haija, M.; Palermo, J.J.; Appakalai, B.N.; Nathan, J.D.; et al. Patient-derived pancreas-on-a-chip to model cystic fibrosis-related disorders. Nat. Commun. 2019, 10, 3124.
- Levink, I.J.M.; Nesteruk, K.; Visser, D.I.; Sieuwerts, A.M.; Fernandes, C.J.C.; Jansen, M.; van Driel, L.; Poley, J.W.; Peppelenbosch, M.P.; Cahen, D.L.; et al. Optimization of pancreatic juice collection: A first step toward biomarker discovery and early detection of pancreatic cancer. Am. J. Gastroenterol. 2020, 115, 2103–2108.
- Molnár, R.; Madácsy, T.; Varga, Á.; Németh, M.; Katona, X.; Görög, M.; Molnár, B.; Fanczal, J.; Rakonczay, Z.; Hegyi, P.; et al. Mouse pancreatic ductal organoid culture as a relevant model to study exocrine pancreatic ion secretion. Lab. Investig. 2020, 100, 84–97.
- Silverman, W.B.; Putnam, P.E.; Orenstein, S.R.; del Rosario, J.F.; Wilson, J.; Kocoshis, S.A. Influence of ERCP on the diagnosis and treatment of pediatric patients with symptomatic pancreatitis and cystic fibrosis: Series of case reports. Gastrointest. Endosc. 1998, 48, 534–536.
- Langron, E.; Prins, S.; Vergani, P. Potentiation of the cystic fibrosis transmembrane conductance regulator by VX-770 involves stabilization of the pre-hydrolytic, O1 state. Br. J. Pharmacol. 2018, 175, 3990–4002.

