2. Bicarbonate Transport in the Exocrine Pancreas
2.1. CFTR Is Indispensable for the Accumulation of Bicarbonate in Pancreatic Juice
In the exocrine pancreas, acinar cells secrete various digestive enzymes in a small volume of isotonic, NaCl- and H
+-rich fluid, after which the ductal epithelium serves to exchange the secreted Cl
− for HCO
3−, to produce an alkaline fluid (pH 8.0–8.5) [
13,
18,
19]. It is estimated that the human exocrine pancreas secretes up to 1 L of pancreatic juice per day [
18,
20,
21]. Apart from delivering digestive enzymes to the proximal small intestine, making it essential for digestion of (macro-)nutrients, pancreatic juice serves to supply base equivalents for neutralization of the gastric acid entering the small intestine from the stomach. In humans, the HCO
3− concentration in pancreatic juice can reach levels of 140 mmol/L [
22,
23]. Similarly high levels of HCO
3− were observed in pancreatic juice of guinea pigs, whereas, in contrast, in mice and rats, HCO
3− levels did not exceed 70 mmol/L [
24,
25,
26,
27,
28].
In humans, the production of pancreatic juice is strongly dependent on postprandial release of the hormone secretin, whereas production is low during fasting. Secretin, through activation of Gα,s-coupled receptors, triggers cyclic AMP (cAMP) production and a consequent protein kinase-mediated phosphorylation and activation of CFTR. In humans, CFTR-mediated, cAMP-dependent ductal fluid secretion is strongly potentiated by parasympathetic, vagal stimulation. Most plausibly, the activation of muscarinic (Gα,q) receptors by acetylcholine triggers K+ efflux through Ca2+-dependent K+ channels, which sustains the negative electrical potential across the luminal plasma membrane driving Cl− and HCO3− efflux through CFTR. In addition to cholinergic neurons, the parasympathetic system comprises neurons that produce vasoactive intestinal polypeptide (VIP), which, as with secretin, activates Gα,s-coupled receptors.
2.2. Molecular Mechanisms of CFTR-Dependent Bicarbonate Transport
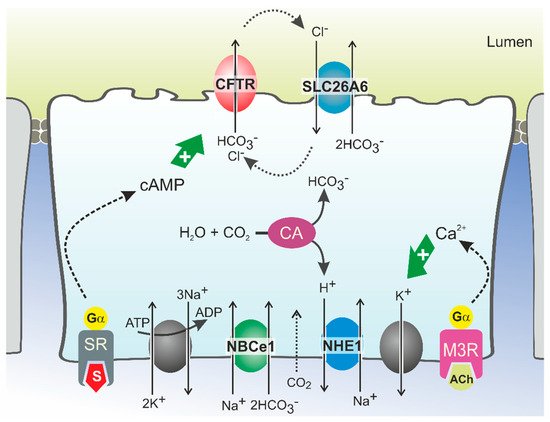
HCO
3− secretion involves the coordinated activity of ion transporters located at the basolateral and luminal membranes of pancreatic ductal cells (PDCs) (
Figure 1). According to current models, which are discussed in detail elsewhere, uptake of HCO
3− from the interstitial space is principally mediated by a Na
+/HCO
3− cotransporter (NBCe1,
SLC4A4) located in the basolateral plasma membrane [
13,
29]. This transport is driven by the steep electrochemical Na
+ gradient across the plasma membrane (generated by Na
+,K
+-ATPase activity) and allows for intracellular accumulation of HCO
3− up to levels well above electrochemical equilibrium. In addition, HCO
3− may be produced intracellularly from carbonic anhydrase-catalyzed hydration of CO
2, which enters the cells through diffusion (
Figure 1). While the protons produced in this reaction are extruded across the basolateral plasma membrane via a Na
+/H
+ exchange mechanism, HCO
3− is secreted across the apical membrane into the ductal lumen via the coordinated activity of the CFTR channel and the Cl
−/HCO
3− exchangers SLC26A6 and, more tentatively, SLC26A3 [
13,
30,
31,
32]. According to this model, which may also be relevant for other HCO
3−-secreting epithelia such as the intestinal and biliary epithelia, CFTR serves to facilitate Cl
−/HCO
3− exchanger-mediated HCO
3− secretion by extruding the Cl
− ions absorbed through the exchangers [
22,
30,
33,
34]. As for CFTR, the activity of Cl
−/HCO
3− exchangers is stimulated by cAMP and Ca
2+ agonists, which implies that their activity is also controlled through neuro-endocrine stimulation of Gα,s- and Gα,q-coupled receptors [
35]. CFTR may not only couple functionally to SLC26A6, but also physically, through binding of its regulatory (R) domain to the sulfate transporter anti-sigma (STAS) domain of SLC26A6 [
36,
37]. This interaction was shown to promote Cl
−/HCO
3− exchange, even in the absence of CFTR activity [
22,
36].
Figure 1. Model of HCO3− secretion in pancreatic ducts. HCO3− secretion is controlled by neuro-endocrine input, chiefly by secretin (S) and acetylcholine (ACh), which increase cellular cAMP and Ca2+ levels, respectively, via stimulation of G protein-coupled receptors. This triggers Na+-coupled HCO3− uptake across the basolateral plasma membrane through NBCe1 and luminal HCO3− efflux through CFTR and SLC26A6. HCO3− may also be accumulated intracellularly via the coordinated activity of carbonic anhydrases (CA) and a proton extrusion mechanism, NHE1. Opening of K+ channels sustains the negative membrane potential which drives (CFTR-mediated) anion efflux. Cl− ions absorbed via SLC26A6 may be recycled to the ductal lumen via CFTR. Ultimately, ductal ion and fluid secretion is driven by ion and electrical gradients that are maintained by Na+,K+-ATP-ase, located in the basolateral membrane. SR: secretin receptor; M3R: muscarinic acetylcholine receptor 3.
Although there is ample evidence for the role of SLC26-type Cl
−/HCO
3− exchangers in ductal HCO
3− secretion, mathematical modeling of the ion motive forces in the ductal epithelium predicted that Cl
−/HCO
3− exchange mechanisms only significantly contribute to cellular HCO
3− extrusion up to luminal HCO
3− levels of circa 70 mmol/L [
29,
38]. Consequently, SLC26-type HCO
3− transport mechanisms may serve to secrete HCO
3− in the proximal segments of the ductal tree, but to reach the final, high HCO
3− levels observed in human pancreatic juice, an alternative transport mechanism must prevail in the distal part. In view of these considerations, it seems plausible that CFTR has a direct role in ductal HCO
3− secretion. The CFTR channel itself was shown to directly mediate cellular HCO
3− transport in studies on both cell models and epithelial tissues, including pancreatic ducts [
39,
40,
41,
42]. It has, however, also been noted that the permeability of the CFTR channel pore for HCO
3− is 4–5 times lower than for Cl
− [
39,
40,
43,
44]. This implies that CFTR can only selectively secrete HCO
3− when very low intracellular Cl
− and comparatively high HCO
3− concentrations are attained. This situation may occur in the distal part of the ductal tree where, upon hormonal stimulation of pancreatic secretion, Cl
− levels in PDCs decrease to values as low as 5 mmol/L [
29]. In these settings, assuming that, concurrently, intracellular HCO
3− is actively accumulated through NBCe1-mediated uptake, it is conceivable that CFTR may function primarily, if not exclusively, as a HCO
3− transporter.
To account for the high HCO
3− and low Cl
− levels found in pancreatic juice, it has been further proposed that the Cl
− over HCO
3− conductance ratio of CFTR is not static but can be lowered to allow selective HCO
3− secretion [
45,
46,
47,
48]. This dynamic regulation of CFTR anion conductance is thought to be mediated by members of the WNK (with no lysine) type of protein kinases [
13,
18,
49,
50]. WNK kinases (the family consists of four members) regulate an array of ion transporters through activation of downstream protein kinases OSR1 (oxidative stress-responsive kinase 1) and the highly homologous SPAK (STE20/SPS1-related proline/alanine-rich kinase) [
51]. The WNK kinases are thought to function as osmotic stress sensors and are activated by a lowering of intracellular Cl
− levels. Notably, WNK1 and WNK4 control renal electrolyte transport, and mutations in either cause hypertension as a result of an unchecked tubular Na
+ re-absorption. In most cases, WNK kinases regulate the activity of ion transport mechanisms by controlling their localization in the plasma membrane.
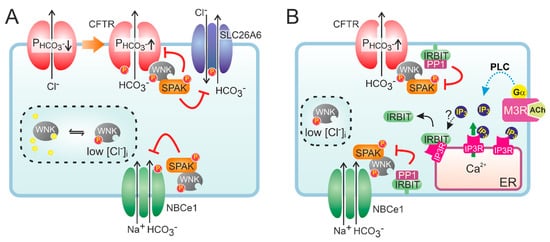
At low extracellular Cl
− concentrations, WNK1-mediated OSR1 and SPAK activation was shown to markedly increase the HCO
3− permeability of CFTR in CFTR-transfected HEK293 cells and in guinea pig PDCs [
52]. Interestingly, WNK1 activation depended on CFTR function as the change in HCO
3− permeability was not observed in cells expressing non-functional, mutant (F508del) CFTR, suggesting CFTR is required for lowering of the intracellular Cl
− concentration. More recently, it was proposed that WNK1 increases the HCO
3− permeability of CFTR by a direct interaction that does not require SPAK activation (
Figure 2) [
53]. WNK kinases are also thought to reduce the surface expression of Cl
−/HCO
3− exchangers of the SLC26A family [
54]. At high luminal HCO
3− levels such as those encountered in the distal ductal tree, this silencing of Cl
−/HCO
3− exchange activity may prevent re-uptake of HCO
3− and, consequently, promote its net secretion. Paradoxically, both WNK1 and WNK4 were also shown to reduce CFTR protein levels at the cell surface and suppress CFTR-mediated anion efflux [
55]. Moreover, WNK kinases may also reduce NBCe1 activity, suggesting that their activation opposes CFTR-mediated HCO
3− secretion [
56]. However, the inhibition imposed by the WNK/SPAK pathway on ductal fluid and HCO
3− secretion is counteracted by the action of the scaffolding protein inositol-1,4,5-trisphosphate (IP
3) receptor-binding protein released with IP
3 (IRBIT) [
56]. Through its PDZ domain, IRBIT co-localizes at the apical and basolateral plasma membrane with CFTR and NBCe1, respectively, and prevents WNK1/SPAK-mediated endocytosis by recruiting a protein phosphatase (PP1) that counters SPAK-mediated phosphorylation of CFTR and NBCe1 [
56,
57,
58]. In addition to its effect on surface expression, IRBIT may also increase the CFTR open channel probability [
59]. Recruitment of IRBIT to these molecular complexes appears to be stimulated through cAMP and Ca
2+ signaling, and IRBIT is required for the synergism observed between these signaling pathways [
60,
61].
Figure 2. Hypothetical model of the role of WNK and IRBIT in ductal HCO3− secretion. (A) Activation of CFTR decreases intracellular Cl−, which relieves the inhibition imposed by Cl− on WNK and stimulates auto-phosphorylation of the protein kinase. Activated WNK binds to CFTR and increases the HCO3− permeability (P) of the channel pore, independent of its protein kinase activity. However, WNK also recruits SPAK, which leads to a phosphorylation-mediated decrease in the cell surface expression of HCO3− transporters, including CFTR. (B) IRBIT is bound to IP3 receptors (IP3R) on cellular Ca2+ stores, e.g., the endoplasmic reticulum (ER). Conceivably, IRBIT is displaced from IP3R upon a phospholipase C (PLC)-mediated increase in IP3 production, after which it translocates to the plasma membrane. Through its PDZ domain, IRBIT anchors to the actin cytoskeleton (not shown) and recruits a protein phosphatase (PP1) to the WNK/SPAK complex. PP1 activity counteracts SPAK-dependent phosphorylation of HCO3− transporters, thus stabilizing their expression at the cell surface. IRBIT does not affect the WNK-dependent but rather the SPAK-independent increase in CFTR HCO3− permeability.