 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Maria Elisa Lopes Pires | + 11276 word(s) | 11276 | 2022-01-04 08:46:10 | | | |
| 2 | Rita Xu | -8686 word(s) | 2590 | 2022-01-14 03:04:26 | | |
Video Upload Options
Sepsis is regarded as one of the main causes of death among the critically ill. Pathogen infection results in a host-mediated pro-inflammatory response to fight infection; as part of this response, significant endogenous reactive oxygen (ROS) and nitrogen species (RNS) production occurs, instigated by a variety of sources, including activated inflammatory cells, such as neutrophils, platelets, and cells from the vascular endothelium. Inflammation can become an inappropriate self-sustaining and expansive process, resulting in sepsis. Patients with sepsis often exhibit loss of aspects for normal vascular homeostatic control, resulting in abnormal coagulation events and development of disseminated intravascular coagulation. Diagnosis and treatment of sepsis remains a significant challenge for health care providers globally. Targeting the drivers of excessive oxidative/nitrosative stress using antioxidant treatments might be a therapeutic option.
1. Introduction
2. Disseminated Intravascular Coagulation in Sepsis
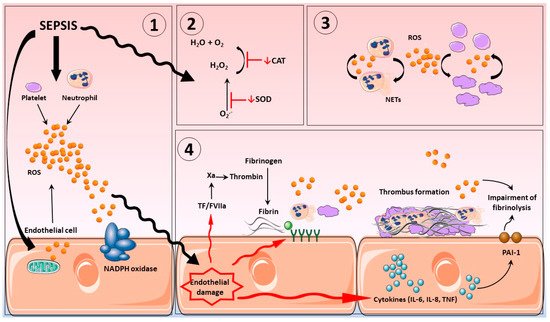
3. Vascular Haemostasis in Sepsis
3.1. Endothelium
3.2. Platelets
3.3. Neutrophils
References
- Bauer, M.; Gerlach, H.; Vogelmann, T.; Preissing, F.; Stiefel, J.; Adam, D. Mortality in Sepsis and Septic Shock in Europe, North America and Australia between 2009 and 2019-Results from a Systematic Review and Meta-Analysis. Crit. Care 2020, 24, 239.
- Dave, M.; Barry, S.; Coulthard, P.; Daniels, R.; Greenwood, M.; Seoudi, N.; Walton, G.; Patel, N. An Evaluation of Sepsis in Dentistry. Br. Dent. J. 2021, 230, 351–357.
- Rhee, C.; Dantes, R.; Epstein, L.; Murphy, D.J.; Seymour, C.W.; Iwashyna, T.J.; Kadri, S.S.; Angus, D.C.; Danner, R.L.; Fiore, A.E.; et al. Incidence and Trends of Sepsis in US Hospitals Using Clinical vs. Claims Data, 2009-2014. JAMA—J. Am. Med. Assoc. 2017, 318, 1241–1249.
- Daniels, R. Survive Sepsis; United Kingdom Sepsis Trust: Birmingham, UK, 2014; ISBN 9780992815509.
- Angus, D.C.; van der Poll, T. Severe Sepsis and Septic Shock. N. Engl. J. Med. 2013, 369, 840–851.
- Cecconi, M.; Evans, L.; Levy, M.; Rhodes, A. Sepsis and Septic Shock. Lancet 2018, 392, 75–87.
- Vincent, J.; Moreno, R.; Takala, J.; de Mendonça, A.; Bruining, H.; Reinhart, C.; Suter, P.; Thijs, L. The SOFA (Sepsis-Related Organ Failure Assessment) Score to Describe Organ Dysfunction/Failure. On Behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med. 1996, 22, 707–710.
- Okabayashi, K.; Wada, H.; Ohta, S.; Shiku, H.; Nobori, T.; Maruyama, K. Hemostatic Markers and the Sepsis-related Organ Failure Assessment Score in Patients with Disseminated Intravascular Coagulation in an Intensive Care Unit. Am. J. Hematol. 2004, 76, 225–229.
- Levi, M.; Cate, H. Ten Disseminated Intravascular Coagulation. N. Engl. J. Med. 1999, 341, 586–592.
- Gando, S.; Levi, M.; Toh, C.H. Disseminated Intravascular Coagulation. Nat. Rev. Dis. Primers 2016, 2, 16037.
- Hoshino, K.; Kitamura, T.; Nakamura, Y.; Irie, Y.; Matsumoto, N.; Kawano, Y.; Ishikura, H. Usefulness of Plasminogen Activator Inhibitor-1 as a Predictive Marker of Mortality in Sepsis. J. Intensive Care 2017, 5, 42.
- Robbie, L.A.; Dummer, S.; Booth, N.A.; Adey, G.D.; Bennett, B. Plasminogen Activator Inhibitor 2 and Urokinase-Type Plasminogen Activator in Plasma and Leucocytes in Patients with Severe Sepsis. Br. J. Haematol. 2000, 109, 342–348.
- Raaphorst, J.; Johan Groeneveld, A.B.; Bossink, A.W.; Erik Hack, C. Early Inhibition of Activated Fibrinolysis Predicts Microbial Infection, Shock and Mortality in Febrile Medical Patients. Thromb. Haemost. 2001, 86, 543–549.
- Lados-Krupa, A.; Konieczynska, M.; Chmiel, A.; Undas, A. Increased Oxidation as an Additional Mechanism Underlying Reduced Clot Permeability and Impaired Fibrinolysis in Type 2 Diabetes. J. Diabetes Res. 2015, 2015, 456189.
- Gando, S.; Saitoh, D.; Ishikura, H.; Ueyama, M.; Otomo, Y.; Oda, S.; Kushimoto, S.; Tanjoh, K.; Mayumi, T.; Ikeda, T.; et al. A Randomized, Controlled, Multicenter Trial of the Effects of Antithrombin on Disseminated Intravascular Coagulation in Patients with Sepsis. Crit. Care 2013, 17, R297.
- Masuda, T.; Shoko, T. Clinical Investigation of the Utility of a Pair of Coagulation-Fibrinolysis Markers for Definite Diagnosis of Sepsis-Induced Disseminated Intravascular Coagulation: A Single-Center, Diagnostic, Prospective, Observational Study. Thromb. Res. 2020, 192, 116–121.
- Matsuda, K.; Kurokawa, M. Underlying Disease and Clinical Phenotypes of Disseminated Intravascular Coagulation. JMA J. 2020, 3, 357–358.
- Ohbe, H.; Yamakawa, K.; Taniguchi, K.; Morita, K.; Matsui, H.; Fushimi, K. Underlying Disorders, Clinical Phenotypes, and Treatment Diversity among Patients with Disseminated Intravascular Coagulation. JMA J. 2020, 3, 321–329.
- Jackson Chornenki, N.L.; Dwivedi, D.J.; Kwong, A.C.; Zamir, N.; Fox-Robichaud, A.E.; Liaw, P.C. Identification of Hemostatic Markers That Define the Pre-DIC State: A Multi-Center Observational Study. J. Thromb. Haemost. 2020, 18, 2524–2531.
- Smith, L. Disseminated Intravascular Coagulation. Semin. Oncol. Nurs. 2021, 37, 151135.
- Yamakawa, K.; Yoshimura, J.; Ito, T.; Hayakawa, M.; Hamasaki, T.; Fujimi, S. External Validation of the Two Newly Proposed Criteria for Assessing Coagulopathy in Sepsis. Thromb. Haemost. 2019, 119, 203–212.
- Naime, A.C.A.; Ganaes, J.O.F.; Lopes-Pires, M.E. Sepsis: The Involvement of Platelets and the Current Treatments. Curr. Mol. Pharmacol. 2018, 11, 261–269.
- Helms, J.; Severac, F.; Merdji, H.; Clere-Jehl, R.; François, B.; Mercier, E.; Quenot, J.P.; Meziani, F. Performances of Disseminated Intravascular Coagulation Scoring Systems in Septic Shock Patients. Ann. Intensive Care 2020, 10, 92.
- Abrams, S.T.; Morton, B.; Alhamdi, Y.; Alsabani, M.; Lane, S.; Welters, I.D.; Wang, G.; Toh, C.H. A Novel Assay for Neutrophil Extracellular Trap Formation Independently Predicts Disseminated Intravascular Coagulation and Mortality in Critically Ill Patients. Am. J. Respir. Crit. Care Med. 2019, 200, 869–880.
- Patel, P.; Walborn, A.; Rondina, M.; Fareed, J.; Hoppensteadt, D. Markers of Inflammation and Infection in Sepsis and Disseminated Intravascular Coagulation. Clin. Appl. Thromb. Hemost. 2019, 25, 1–6.
- Yang, X.; Cheng, X.; Tang, Y.; Qiu, X.; Wang, Y.; Kang, H.; Wu, J.; Wang, Z.; Liu, Y.; Chen, F.; et al. Bacterial Endotoxin Activates the Coagulation Cascade through Gasdermin D-Dependent Phosphatidylserine Exposure. Immunity 2019, 51, 983–996.e6.
- Kinasewitz, G.T.; Yan, S.B.; Basson, B.; Comp, P.; Russell, J.A.; Cariou, A.; Um, S.L.; Utterback, B.; Laterre, P.F.; Dhainaut, J.F. Universal Changes in Biomarkers of Coagulation and Inflammation Occur in Patients with Severe Sepsis, Regardless of Causative Micro-Organism . Crit. Care 2004, 8, 82–90.
- Levi, M.; van der Poll, T. Coagulation and Sepsis. Thromb. Res. 2017, 149, 38–44.
- Scully, M.; Levi, M. How We Manage Haemostasis during Sepsis. Br. J. Haematol. 2019, 185, 209–218.
- Nieuwland, R.; Gardiner, C.; Dignat-George, F.; Mullier, F.; Mackman, N.; Woodhams, B.; Thaler, J. Toward Standardization of Assays Measuring Extracellular Vesicle-Associated Tissue Factor Activity. J. Thromb. Haemost. 2019, 17, 1261–1264.
- Kay, J.G.; Grinstein, S. Phosphatidylserine-Mediated Cellular Signaling. Adv. Exp. Med. Biol. 2013, 991, 177–193.
- Delabranche, X.; Boisramé-Helms, J.; Asfar, P.; Berger, A.; Mootien, Y.; Lavigne, T.; Grunebaum, L.; Lanza, F.; Gachet, C.; Freyssinet, J.M.; et al. Microparticles Are New Biomarkers of Septic Shock-Induced Disseminated Intravascular Coagulopathy. Intensive Care Med. 2013, 39, 1695–1703.
- Matsumoto, H.; Yamakawa, K.; Ogura, H.; Koh, T.; Matsumoto, N.; Shimazu, T. Enhanced Expression of Cell-Specific Surface Antigens on Endothelial Microparticles in Sepsis-Induced Disseminated Intravascular Coagulation. Shock 2015, 43, 443–449.
- Walborn, A.; Rondina, M.; Mosier, M.; Fareed, J.; Hoppensteadt, D. Endothelial Dysfunction Is Associated with Mortality and Severity of Coagulopathy in Patients with Sepsis and Disseminated Intravascular Coagulation. Clin. Appl. Thromb. Hemost. 2019, 25, 1–9.
- VanTeeffelen, J.W.; Brands, J.; Stroes, E.S.; Vink, H. Endothelial Glycocalyx: Sweet Shield of Blood Vessels. Trends Cardiovasc. Med. 2007, 17, 101–105.
- Lupu, F.; Kinasewitz, G.; Dormer, K. The Role of Endothelial Shear Stress on Haemodynamics, Inflammation, Coagulation and Glycocalyx during Sepsis. J. Cell. Mol. Med. 2020, 24, 12258–12271.
- Sampei, S.; Okada, H.; Tomita, H.; Takada, C.; Suzuki, K.; Kinoshita, T.; Kobayashi, R.; Fukuda, H.; Kawasaki, Y.; Nishio, A.; et al. Endothelial Glycocalyx Disorders May Be Associated With Extended Inflammation During Endotoxemia in a Diabetic Mouse Model. Front. Cell Dev. Biol. 2021, 9, 623582.
- Kushimoto, S.; Abe, T.; Ogura, H.; Shiraishi, A.; Saitoh, D.; Fujishima, S.; Mayumi, T.; Hifumi, T.; Shiino, Y.; Nakada, T.-A.; et al. Impact of Blood Glucose Abnormalities on Outcomes and Disease Severity in Patients with Severe Sepsis: An Analysis from a Multicenter, Prospective Survey of Severe Sepsis. PLoS ONE 2020, 15, e0229919.
- Iba, T.; Connors, J.M.; Nagaoka, I.; Levy, J.H. Recent Advances in the Research and Management of Sepsis-Associated DIC. Int. J. Hematol. 2021, 113, 24–33.
- Hou, P.C.; Filbin, M.R.; Wang, H.; Ngo, L.; Aird, W.C.; Shapiro, N.I.; Wang, H.; Huang, D.T.; Angus, D.C.; Kellum, J.A.; et al. Endothelial Permeability and Hemostasis in Septic Shock: Results From the ProCESS Trial. Chest 2017, 152, 22–31.
- Portier, I.; Campbell, R.A. Role of Platelets in Detection and Regulation of Infection. Arterioscler. Thromb. Vasc. Biol. 2020, 41, 70–78.
- Ma, R.; Xie, R.; Yu, C.; Si, Y.; Wu, X.; Zhao, L.; Yao, Z.; Fang, S.; Chen, H.; Novakovic, V.; et al. Phosphatidylserine-Mediated Platelet Clearance by Endothelium Decreases Platelet Aggregates and Procoagulant Activity in Sepsis. Sci. Rep. 2017, 7, 4978.
- Puskarich, M.A.; Cornelius, D.C.; Bandyopadhyay, S.; McCalmon, M.; Tramel, R.; Dale, W.D.; Jones, A.E. Phosphatidylserine Expressing Platelet Microparticle Levels at Hospital Presentation Are Decreased in Sepsis Non-Survivors and Correlate with Thrombocytopenia. Thromb. Res. 2018, 168, 138–144.
- Tsirigotis, P.; Chondropoulos, S.; Frantzeskaki, F.; Stamouli, M.; Gkirkas, K.; Bartzeliotou, A.; Papanikolaou, N.; Atta, M.; Papassotiriou, I.; Dimitriadis, G.; et al. Thrombocytopenia in Critically Ill Patients with Severe Sepsis/Septic Shock: Prognostic Value and Association with a Distinct Serum Cytokine Profile. J. Crit. Care 2016, 32, 9–15.
- Lopes-Pires, M.E.; Naime, A.C.A.; Cardelli, N.J.A.; Anjos, D.J.; Antunes, E.; Marcondes, S. PKC and AKT Modulate CGMP/PKG Signaling Pathway on Platelet Aggregation in Experimental Sepsis. PLoS ONE 2015, 10, e0137901.
- Naime, A.C.A.; Bonfitto, P.H.L.; Solon, C.; Lopes-Pires, M.E.; Anhê, G.F.; Antunes, E.; Marcondes, S. Tumor Necrosis Factor Alpha Has a Crucial Role in Increased Reactive Oxygen Species Production in Platelets of Mice Injected with Lipopolysaccharide. Platelets 2019, 30, 1047–1052.
- Laursen, M.A.; Larsen, J.B.; Larsen, K.M.; Hvas, A.M. Platelet Function in Patients with Septic Shock. Thromb. Res. 2020, 185, 33–42.
- Bardoel, B.W.; Kenny, E.F.; Sollberger, G.; Zychlinsky, A. The Balancing Act of Neutrophils. Cell Host Microbe 2014, 15, 526–536.
- Metzler, K.D.; Goosmann, C.; Lubojemska, A.; Zychlinsky, A.; Papayannopoulos, V. Myeloperoxidase-Containing Complex Regulates Neutrophil Elastase Release and Actin Dynamics during NETosis. Cell Rep. 2014, 8, 883–896.
- Xie, T.; Duan, Z.; Sun, S.; Chu, C.; Ding, W. β-Lactams Modulate Neutrophil Extracellular Traps Formation Mediated by MTOR Signaling Pathway. Biochem. Biophys. Res. Commun. 2021, 534, 408–414.
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.V.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-Induced NETosis Is a Dynamic Process Involving Neutrophil Multitasking in Vivo. Nat. Med. 2012, 18, 1386–1393.
- Shimizu, M.; Konishi, A.; Nomura, S. Examination of Biomarker Expressions in Sepsis-Related DIC Patients. Int. J. Gen. Med. 2018, 11, 353–361.
- Jiao, Y.; Li, W.; Wang, W.; Tong, X.; Xia, R.; Fan, J.; Du, J.; Zhang, C.; Shi, X. Platelet-Derived Exosomes Promote Neutrophil Extracellular Trap Formation during Septic Shock. Crit. Care 2020, 24, 380.
- Rodrigues, D.A.S.; Prestes, E.B.; Gama, A.M.S.; de Souza Silva, L.; Pinheiro, A.A.S.; Ribeiro, J.M.C.; Campos, R.M.P.; Pimentel-Coelho, P.M.; de Souza, H.S.; Dicko, A.; et al. CXCR4 and MIF Are Required for Neutrophil Extracellular Trap Release Triggered by Plasmodium-Infected Erythrocytes. PLoS Pathog. 2020, 16, e1008230.
- Colón, D.F.; Wanderley, C.W.; Franchin, M.; Silva, C.M.; Hiroki, C.H.; Castanheira, F.V.S.; Donate, P.B.; Lopes, A.H.; Volpon, L.C.; Kavaguti, S.K.; et al. Neutrophil Extracellular Traps (NETs) Exacerbate Severity of Infant Sepsis. Crit. Care 2019, 23, 113.
- Huang, H.; Tohme, S.; Al-Khafaji, A.B.; Tai, S.; Loughran, P.; Chen, L.; Wang, S.; Kim, J.; Billiar, T.; Wang, Y.; et al. Damage-Associated Molecular Pattern-Activated Neutrophil Extracellular Trap Exacerbates Sterile Inflammatory Liver Injury. Hepatology 2015, 62, 600–614.
- Shrestha, B.; Ito, T.; Kakuuchi, M.; Totoki, T.; Nagasato, T.; Yamamoto, M.; Maruyama, I. Recombinant Thrombomodulin Suppresses Histone-Induced Neutrophil Extracellular Trap Formation. Front. Immunol. 2019, 10, 2535.
- Okeke, E.B.; Louttit, C.; Fry, C.; Najafabadi, A.H.; Han, K.; Nemzek, J.; Moon, J.J. Inhibition of Neutrophil Elastase Prevents Neutrophil Extracellular Trap Formation and Rescues Mice from Endotoxic Shock. Biomaterials 2020, 238, 119836.
- Kumar, S.; Gupta, E.; Kaushik, S.; Srivastava, V.K.; Saxena, J.; Mehta, S.; Jyoti, A. Quantification of NETs Formation in Neutrophil and Its Correlation with the Severity of Sepsis and Organ Dysfunction. Clin. Chim. Acta 2019, 495, 606–610.
- Chirivi, R.G.S.; van Rosmalen, J.W.G.; van der Linden, M.; Euler, M.; Schmets, G.; Bogatkevich, G.; Kambas, K.; Hahn, J.; Braster, Q.; Soehnlein, O.; et al. Therapeutic ACPA Inhibits NET Formation: A Potential Therapy for Neutrophil-Mediated Inflammatory Diseases. Cell. Mol. Immunol. 2020, 18, 1528–1544.
- Locke, M.; Francis, R.J.; Tsaousi, E.; Longstaff, C. Fibrinogen Protects Neutrophils from the Cytotoxic Effects of Histones and Delays Neutrophil Extracellular Trap Formation Induced by Ionomycin. Sci. Rep. 2020, 10, 11694.
- Ode, Y.; Aziz, M.; Jin, H.; Arif, A.; Nicastro, J.G.; Wang, P. Cold-Inducible RNA-Binding Protein Induces Neutrophil Extracellular Traps in the Lungs during Sepsis. Sci. Rep. 2019, 9, 6252.
- Levi, M.; Scully, M.; Singer, M. The Role of ADAMTS-13 in the Coagulopathy of Sepsis. J. Thromb. Haemost. 2018, 16, 646–651.
- Hakkim, A.; Fürnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of Neutrophil Extracellular Trap Degradation Is Associated with Lupus Nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818.
- Müller, F.; Mutch, N.J.; Schenk, W.A.; Smith, S.A.; Esterl, L.; Spronk, H.M.; Schmidbauer, S.; Gahl, W.A.; Morrissey, J.H.; Renné, T. Platelet Polyphosphates Are Proinflammatory and Procoagulant Mediators In Vivo. Cell 2009, 139, 1143–1156.
- Smith, S.A.; Mutch, N.J.; Baskar, D.; Rohloff, P.; Docampo, R.; Morrissey, J.H. Polyphosphate Modulates Blood Coagulation and Fibrinolysis. Proc. Natl. Acad. Sci. USA 2006, 103, 903–908.

