Sepsis is regarded as one of the main causes of death among the critically ill. Pathogen infection results in a host-mediated pro-inflammatory response to fight infection; as part of this response, significant endogenous reactive oxygen (ROS) and nitrogen species (RNS) production occurs, instigated by a variety of sources, including activated inflammatory cells, such as neutrophils, platelets, and cells from the vascular endothelium. Inflammation can become an inappropriate self-sustaining and expansive process, resulting in sepsis. Patients with sepsis often exhibit loss of aspects for normal vascular homeostatic control, resulting in abnormal coagulation events and development of disseminated intravascular coagulation. Diagnosis and treatment of sepsis remains a significant challenge for health care providers globally. Targeting the drivers of excessive oxidative/nitrosative stress using antioxidant treatments might be a therapeutic option. This review focuses on the association between excessive oxidative/nitrosative stress, a common feature in sepsis, and loss of homeostatic control at the level of the vasculature. Literature relating to potential antioxidants is also described.
- sepsis
- oxidative stress
- nitric oxide
- netosis
- platelets
- clotting dysfunction
1. Introduction
2. Disseminated Intravascular Coagulation in Sepsis
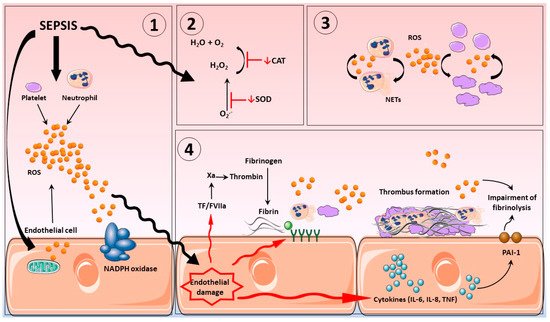
3. Vascular Haemostasis in Sepsis
3.1. Endothelium
3.2. Platelets
3.3. Neutrophils
- Oxidative and nitrosative stress
Reactive oxygen ROS are chemical species encompassing free radicals and related oxygen containing species. The most encountered inorganic ROS include: the superoxide radical anion (O2•-), the hydroxyl radical (OH•), hydrogen peroxide (H2O2), and hypochlorous acid (HOCl) [59–61] . Most ROS production is purposeful allowing for utilizations of oxygen for aerobic metabolism and biosynthesis. These processes are highly regulated to limit any adverse consequences related to the inappropriate activation of oxygen to more reactive forms. Should this control be under-mined or lost excessive ROS production can affect redox regulated cell-signalling responses leading to aberrant stress responses as seen in sepsis. To this end, over production of O2•- and H2O2, both key signalling species, can lead to redox-regulated pro-inflammatory transcription factor over activation and significant and inappropriate pro-inflammatory responses. The production of directly damaging ROS including OH• and HOCl can also result in damage and dysfunction to an array of biomolecules including lipids, proteins and nucleic acids and the production of toxic end-products such as aldehydes and protein carbonyls. Moreover, interactions between relatively innocuous species such as O2•- with nitric oxide (NO) in equimolar proportions results in the production of peroxynitrite (ONOO-) which, although not a free radical, is nevertheless a very aggressive species capable of inflicting damage similar to that ascribed to OH•. Furthermore, ONOO- is capable of reacting with proteins and peptides thereby causing functional changes; modifications include s-nitrosylation, glutathionylation and tyrosine nitration. NO and ONOO- are described as reactive nitrogen species (RNS); other RNS include nitroxyl (HNO), nitrosonium cation (NO+), S-nitrosothiols (RSNOs), NO2- (nitrite) and dinitrosyl iron complexes, excluding NO3 [62,63]. Under normal conditions, endothelial cells generate NO through eNOS, influencing cGMP levels, relaxing vascular smooth muscle thereby promoting the vessel dilation. NO is also a known inhibitor of platelet activity through sGC- cGMP- PKG pathway, following different mechanisms: i) PKG reduces intraplatelet Ca+2 levels inhibiting platelet shape change and consequently, inhibiting the release of mediators involved in platelet aggregation; ii) PKG promotes the phosphorylation of TXA2 receptor, suppressing the effects of the platelet agonist; iii) platelet aggregation is inhibited by the synergic effect of cGMP and cAMP; iv) cGMP inhibits PI3K, which is responsible for the activation of integrin αIIbβ3, a transmembrane glycoprotein signalling receptors essential for normal platelet function. NO donors also have been shown to inhibit platelet aggregation independently of sGC [68].
Additionally, it is well reported that haemoglobin (Hb) is capable of binding to NO and its metabolites. S-nitrosothiols and dinitrosyl iron complexes bind to the heme in Hb, working as a NO store, preventing NO oxidation. Moreover, despite the fact that endothelial NO synthesis in venous circulation is disabled due a low concentration of oxygen, the Hb stores of NO guarantee that NO is available to the venous circulation [69,70]. Appropriate regulation of NO generation and distribution is therefore fundamental for the maintenance of vascular tone and normal blood flow, regulating platelet and leukocyte adhesion to endothelium and ultimately the distribution of oxygen and nutrients to the body [71–73]. Additionally, heme is converted to carbon monoxide (CO), free iron and bilirubin through heme oxygenase (HO-1) action. Although CO has been reported to induce anti-inflammatory cytokines and downregulating pro-inflammatory cytokines release (reviewed by Ryter, 2016), its pro-coagulant and anti-fibrinolysis activities (reviewed by Nielsen, 2014) might be substantially involved in the induction of generalized coagulation in sepsis.
Normally, the collateral effects of ROS production are limited due to an armory of protective strategies chiefly facilitated by diverse forms of antioxidant protection and efficient removal and repair mechanisms. However, under circumstances such as excessive inflammation, traumatic injury and cell/ tissue ischaemia, excessive levels of ROS formation can occur to the extent that endogenous protection becomes overwhelmed, a scenario often observed in the critically ill and particularly so during sepsis. Indeed, there is
a well-established literature demonstrating oxidative and nitroasive modification/damage of biomolecules in the critically ill [74–78]. In addition, the production of DAMPs (damage-associated molecular patterns) and PAMPs (pathogen associated molecular patterns) and subsequent binding to activation of TLRs (toll like receptors), induces increased ROS release by an array of cells, including endothelial cells, platelets and neutrophils [79]. Such activation further promotes additional ROS production by these cells, creating a self-sustaining and ever-expanding ROS activation system, which further negatively impacts patient clinical presentation [80–83].
As for the systems which generate ROS in sepsis, these are complex including, NADPH oxidase (NOX) and dual oxidase enzymes (DuOX); mitochondrial respiration and dysfunction; the activities of cyclooxygenases and lipoxygenases; xanthine oxidoreductases (XOR); the effects of ischaemia reperfusion injury; NO production by NOS enzymes; loss of homeostatic control for iron recycling allowing for production of directly damaging ROS. See table 1 for specific details.
Table 1. Main reactive oxygen and nitric species producers.
|
Enzyme |
Mechanism |
Reference |
|
Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX1-5; DUOX1, 2) |
Conversion of O2 to O2•-, NADPH acts as an electron donor. NOX1-4 provide constitutive activity, which is dependent on subunits NOXO1, p47phox or p22 phox phosphorylation. Further rearrangement of the subunit complexes p40phox, p67phox and Rac from the cytosol to the membrane al-lows for transfer of electrons from the substrate to O2. NOX5 and Duox activation are calcium-dependent. |
[84–86] |
|
Mitochondrial respiration chain |
Oxygen acts the terminal electron acceptor of the respiratory chain. The process involves a 4 electrons reduction of oxygen to water, which can occur in the outer membrane, in the inner membrane or within the matrix. ROS including O2•-, H2O2 and OH• are produced as intermediates in this ongoing process. Around 1% of O2•- exits mitochondria as a physiological process under steady state conditions. Hyperoxia and hypoxia/ reperfusion both augment O2•- release greatly with the potential for direct effects on cellular redox state and signalling and also the conversion to more damaging species through iron catalysis (Fenton reaction). |
[87–91] |
|
Cyclooxygenase and Lipoxygenase |
These enzymes metabolize arachidonic acid (AA) to form prostaglandins, thromboxane and leukotrienes. The enzymic addition of oxygen as occurs in these processes involves ROS generation with the potential for collateral effects. |
[92,93] |
|
Xanthine, oxidoreductase (XO), dehydrogenase oxidase (XDH) |
Rate limiting enzymes responsible for the conversion of hypoxanthine and xanthine to uric acid in the last stages of purine catabolism. XDH catalyses these process utilising NAD+ as a cofactor. XDH can be readily converted to XO by hyperoxia, the effects of ischaemia/ reperfusion or by limited proteolysis. XO catalyses the same reaction but uses oxygen as a co-factor rather than NAD+, consequently, O2•- and H2O2 are generated as by products and thus influence an array of ROS related dysfunctions. |
[94–96] |
|
Nitric oxide synthase (NOS) NOS1 or nNOS (neuronal), NOS2 or iNOS (inducible), and NOS3 or eNOS (endothelial) |
Enzymatic production of NO and regulation of vascular tone. Use of l-arginine and O2 as substrates and nicotinamide-adenine-dinucleotide phosphate (NADPH), flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN), and (6R)5,6,7,8-tetrahydrobiopterin (BH4) as reduced cofactors. |
[96] |
- The Role of oxidative and nitrosative stress in sepsis related haemostasis
5.1. Glycocalyx
The glycocalyx of the endothelium is an intravascular lubricant layer, composed of membrane-binding domains and plasma proteins that separates circulating blood from vessel walls. It is a major contributor to cardiovascular homeostasis, controlling thrombus development, vascular permeability together with the provision of anti-inflammatory and antioxidant defenses [97,98]. Severe inflammation promotes glycocalyx shedding, altering structure and compromising function [97]. Oxidative stress is thought to be a major contributor for this impairment, which ultimately leads to the synthesis and exposure of adhesion molecules and subsequently the influx of leukocytes and platelets to the vascular bed [99,100]. Indeed, it has been reported that in diseases where oxidative stress plays an important role, such as sepsis or post-cardiac arrest syndrome, shedding of glycocalyx structures was apparent (see figure 1) [101].
5.1.1. Mitochondria
Mitochondria are the principal site of ROS generation in endothelial cells both in health and during sepsis [102] and numerous studies have reported the impacts of altered mitochondrial activity on the function of blood vessel. For instance, in human coronary arterioles, rotenone and myxotgiazol, inhibitors of mitochondrial complex I and III respectively, mitigated flow-induced dilatation associated with O2•- and H2O2 release; whereas, apocynin, an NADPH oxidase inhibitor, did not affect the increase of ROS generation induced by shear stress [103]. Additionally, Lowes et al showed that human umbilical vein endothelial cell incubated with LPS, promoted elevated ROS generation, lowered mitochondrial membrane potential and increased the release of cytokines IL-1b, IL-6, IL-8, IL-10. These effects were all abrogated by MitoQ, a mitochondrial targeted antioxidant [104]. MitoTEMPO, another mitochondrial superoxide scavenger, also ameliorated organ dysfunction and improved the survival rates in a murine model of sepsis involving CLP model [105] Additionally, the antioxidant protein paraoxonase-2 (PON2) is involved in the control of oxidative stress, reduction of inflammation and protection against atherosclerosis. Furthermore, Altenhöfer et al have demonstrated that PON2 decreased O2•- generation from endothelial cells of human mitochondrial complex I and complex III at the inner mitochondrial membrane, supposedly by acting on coenzyme Q10 (CoQ) [106]. In addition, Ebert et al showed that PON2- knockout mice presented loss of redox homeostasis; endothelial cells abnormalities with an increase of tissue factor activity; reduction of coagulation times and increased platelet activity [107].
5.1.2. NADPH oxidase
This multisubunit enzyme is also reported to promote ROS generation in the endothelium. Wu et all showed in microvascular endothelial cells when stimulated with LPS, that the ROS scavenger ascorbate abrogated NOX1 activity, p47phox expression and consequently, decreased ROS production [108]. The upregulation of NOX1 by LPS, TNF-α and IL-1α has also shown to induce mitochondrial O2•- generation in pig pulmonary arteries [109].
During sepsis, the endothelium generates high levels of tissue factor pathway inhibitor (TFPI) together with NO and prostacyclin in order to maintain an antithrombotic capacity. However, in sepsis, endothelium anticoagulant factors such as TFPI, do not operate appropriately, thereby, allowing leukocyte and platelet adhesion and consequently the release of tissue factor and the formation of microthrombi [81,106]. Interestingly, incubation of endothelial cells with xanthine/xanthine oxidase a potent source of O2•- and H2O2, inhibits TFPI [110].
5.1.3. iNOS and NO
The glycocalyx can also release toxic levels of NO via iNOS activity, which causes hypotension and circulatory failure, subsequent disruption of oxygen distribution impairment of the endothelial barrier system and damage to various organs [73,111]. Additionally, greatly elevated levels of NO promote a substantial inhibition of platelet activity, leading to an extensive bleeding time, hemorrhage and potentially death [112,113]. Furthermore, in addition to endothelial cells, neutrophils also contain iNOS, which further contributes to the NO pool in sepsis and reduces interaction between neutrophils and the endothelium [114]. In this regard, a study using a cecal ligation and puncture (CLP) model of sepsis in rodents showed that increases in NO production inhibited neutrophil rolling and consequently, abolished adhesion to the endothelium. Importantly treatment with aminoguanidine, an NO inhibitor, was able to restore neutrophil function and decreased mortality [115]. Another relevant pathophysiological RNS species is ONOO-, which is formed by the reaction between equimolar levels of O2•- and NO, this directly damaging species when formed is as-sociated with cytotoxic effects and tissue damage [73,111,116].
5.2. Platelets
Platelets also play a key role in ROS production during sepsis with both external and internal ROS production reported to modulate platelet activity through the integrin αIIbβ3 (fibrinogen receptor), GVI (collagen receptor) and GPIbα (von Willebrand factor receptor) [117,118]. As such, platelets incubated with LPS from Chlamydia pneumoniae, Proteus mirabilis or Escherichia coli all demonstrated elevated levels of ROS generation. In addition, LPS induced ROS generation by platelets, increased platelet- fibrinogen binding and P-selectin exposure, all of which was abrogated by superoxide dismutase and catalase [102,117]. Moreover, mice pre-treated with the antioxidant N-acetylcysteine (NAC) prior to injection with LPS reestablished normal levels of platelet ROS production and aggregation [119].
One of the main roles of platelets in sepsis is to promote the activation and migration of neutrophils to the sites of tissue injury and to stimulate neutrophil NETs release and ROS generation [120,121]. Additionally, both in experimental sepsis and in patients recovering from septic shock, neutrophils also generate extensive levels of production ROS via NADPH oxidase (NOX2), independent of any platelet stimulus. Importantly, neutrophils surface receptors, such as integrin, Fc receptors and members of G-protein-coupled receptors family have all been shown to promote the stimulation of neutrophil- NADPH oxidase activation [122].
It is well known that platelets contain NADPH oxidases, more specifically NOX1, NOX2 and NOX4. However, any role for NOXs in platelet signalling remains somewhat contradictory [123–126]. Some studies have reported a fundamental role in experimental platelet ROS production. Whilst others have shown that endotoxemia in rats increased TNF-α levels and promoted platelet- NADPH oxidase activity, via cGMP-PKG and PKC-p47phox signalling pathways [119,127,128]. NADPH oxidase (NOX2) activity within neutrophils and specifically ROS production has been suggested to stimulate NETosis, as seen in both human and experimental sepsis, [99,129]. However, this assertion is a subject of ongoing debate, as other studies have shown that inhibition of neutrophil NADPH oxidase did not affect NETs release [130,131].
Moreover, in patients with sepsis, soluble plasma factor induced uncoupling of plate-let mitochondria raises respiratory capacity, a feature that was more intense in non-survivors. In addition, platelet mitochondria function was reported to be associated with organ failure and elevated lactate levels [132,133].
- Summary
This review provides some evidence linking aspects of oxidative/ nitroasive stress, and the onset and establishment of hemostasis in sepsis. Abnormal coagulation events including DIC impair tissue prefusion which may ultimately result in multiple organ dysfunction and death. The combination of greatly elevated levels of ROS and RNS production resulting from an overstimulation of the inflammatory response beyond the limits of homeostatic control, in part due to depletion of finite antioxidant reserves, results in a pro-oxidant environment. Under these circumstances redox based stress responses become detrimental with an array of negative impacts including the over-stimulation of coagulation and further expansive inflammatory systems activation; endothelium dysfunction, platelet and neutrophil activation including the formation of neutrophil NETs, all contribute to these responses.
Diagnosis and treatment of sepsis remains a significant challenge for health care providers globally, gaining greater insights into key aspects of complicated proinflammatory processes that ensue during onset and progression of disease remain a priority. Targeting the drivers of excessive oxidative/nitrosative stress using antioxidant treatments is an obvious therapeutic avenue. However, whilst beneficial responses can be demonstrated for an array of adverse endpoints including clotting dysfunction using in vitro and in vivo models, clinical trials have to date been somewhat disappointing; a more nuanced approach may offer a way forward. In this regard, the advent of antioxidants which are specific for key compartments/organelles such as mitochondria, combinational approaches to operative in differing extracellular and intracellular compartments, prophylactic usage in at risk groups, and or the timing and or duration of use may provide some measure of success (see Figure 2 and Table 2).
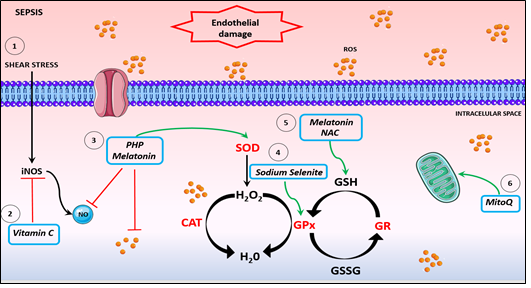
Figure 2. Potential antioxidants therapies. 1. The inflammatory scenario of sepsis induces shear stress, causing endothelium damage and activation of iNOS, leading to a NO boosting. 2. Vitamin C is an antioxidant acting on iNOS inhibition expression, improving microvascular dysfunction and ameliorating hypotension. 3. The compound PHP and melatonin sequestrate NO and promotes SOD activation. 4. Sodium selenite promotes increase of GPx activity. 5. NAC and melatonin restore GSH levels and inhibits platelet and neutrophil dysfunction. 6. MitoQ enhances mitochondrial respiration and restores mitochondrial dysfunction. NO: Nitric oxide. CAT: catalase. SOD: superoxide dismutase. GPx: glutathione peroxidase. GR: glutathione reductase.
Table 2. Potential antioxidants therapies.
|
Therapy |
Mechanism |
Positive effect |
Why is not it been clinically used? |
|
Vitamin C |
Potent ROS scavenging antioxidant agent [134] |
Septic shock patients treated with ANON®, an antioxidant-enriched concentrated liquid diet with high concentrations of vitamin C and E, demonstrated a restoration of vitamin C radical levels in serum and a reduction in MOF [135]. Septic animals treated with vitamin C showed improvement of microvascular dysfunction and microvascular permeability barrier integrity, inhibition of iNOS expression and ameliorated hypotension [83,108,134,136]. The vasodilatation and reduction of vitamin C plasma concentration after low doses of LPS administration in healthy volunteers were reversed by co-administration of vitamin C [137]. |
Limited clinical trials. |
|
|
|
|
|
|
Seleniun |
A micronutrient fundamental for GPx synthesis [138,139] |
The administration of high levels of sodium selenite, intravenously showed an increase of blood selenium concentration and GPx activity and significantly decreased mortality of septic patients with DIC [140]. |
Seleniun decreased the infection in nonseptic patients only. Clinical trials did not show any improvement in outcomes in a general septic patient population [141] |
|
N-acetylcysteine (NAC) |
Antioxidant able to restore the levels of GSH in the cells and also acts as an anti-inflammatory agent [142] |
Conflicting results: Some studies showed that NAC did not im-prove outcome for patients or affect levels of cytokines release [151]. NAC can worsen also organ failure [152]. Findings need to be confirmed in larger clinical trials |
|
|
MitoQ |
Targets mitochondrial dysfunction [110][110] |
Endotoxemic rats that received MitoQ by i.v. administration demonstrated enhancement of mitochondria respiration, decreased levels of oxidative stress and IL-6 and showed improved organ dysfunction [152,153]. |
There is no data from human studies |
|
Superoxide dismutase (SOD) |
Converts superoxide radical into hydrogen peroxide and molecular oxygen (O2), while the catalase and peroxidases convert hydrogen peroxide into water [155,156] |
The M40401 SOD mimetic restored vascular reactivity, regulated arterial pressure and decreased mortality levels of rats infected with E. coli [157] |
There is no data from human studies |
|
Nitric oxide scavenger |
The compound pyridoxylated haemoglobin polyoxyethylene (PHP) is a chemically altered human-derived hemoglobin used as NO scavenger and SOD mimetic [158]. |
In Pseudomona aeruginosa sepsis model in sheep, infusion of PHP for 48h restored a low mean arterial pressure and improved the systemic vascular resistance [159,160]. In phase I/II clinical trials, PHP increased blood pressure and diminished catecholamine requirement [161]; in a phase III trial with 377 patients, PHP reduced the necessity use of vasopressor use [162]. |
Although some positive results, after 28 days of therapy with PHP, there was no benefit and indeed mortality rates increased; with a SOFA score higher than 13 [126]. |
|
Melatonin |
Secreted during the night, melatonin is a hormone produced by pineal gland. It possesses anti-inflammatory properties and in addition demonstrates antioxidant functions acting as both a ROS and RNS scavenger [127]. |
In septic rats induced by CLP, administration of melatonin improved organ injury and effect that was ascribed to the capacity of melatonin enhance GSH levels and to inhibit neutrophil aggregation [164]. In a placebo-controlled study with 12 healthy volunteers, the group that received melatonin before LPS, showed lower levels of inflammatory markers and oxidative stress, compared with the saline control group [129,130]. |
Lack of clinical trials. |
Funding: This study was supported by British Heart Foundation and Coordination for the Improvement of Personnel Higher Education Personnel (CAPES).
Conflicts of Interest: The authors declare no conflict of interest.
References
- Bauer, M.; Gerlach, H.; Vogelmann, T.; Preissing, F.; Stiefel, J.; Adam, D. Mortality in Sepsis and Septic Shock in Europe, North America and Australia between 2009 and 2019-Results from a Systematic Review and Meta-Analysis. Critical Care 2020, 24.
- Dave, M.; Barry, S.; Coulthard, P.; Daniels, R.; Greenwood, M.; Seoudi, N.; Walton, G.; Patel, N. An Evaluation of Sepsis in Dentistry. British Dental Journal 2021, 230, 351–357, doi:10.1038/s41415-021-2724-6.
- Rhee, C.; Dantes, R.; Epstein, L.; Murphy, D.J.; Seymour, C.W.; Iwashyna, T.J.; Kadri, S.S.; Angus, D.C.; Danner, R.L.; Fiore, A.E.; et al. Incidence and Trends of Sepsis in US Hospitals Using Clinical vs Claims Data, 2009-2014. JAMA - Journal of the American Medical Association 2017, 318, 1241–1249.
- Daniels, Rob. Survive Sepsis.; UK Sepsis Trust, 2014; ISBN 9780992815509.
- Angus, D.C.; van der Poll, T. Severe Sepsis and Septic Shock. The New England Journal of Medicine 2013, 369, 840–851, doi:10.1007/978-81-322-0535-7_88.
- Cecconi, M.; Evans, L.; Levy, M.; Rhodes, A. Sepsis and Septic Shock. The Lancet 2018, 392, 75–87, doi:10.1016/S0140-6736(18)30696-2.
- Vincent, J.; Moreno, R.; Takala, J.; de Mendonça, A.; Bruining, H.; Reinhart, C.; Suter, P.; Thijs, L. The SOFA (Sepsis-Related Organ Failure Assessment) Score to Describe Organ Dysfunction/Failure. On Behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med 1996, 22, 707–710.
- Okabayashi, K.; Wada, H.; Ohta, S.; Shiku, H.; Nobori, T.; Maruyama, K. Hemostatic Markers and the Sepsis‐related Organ Failure Assessment Score in Patients with Disseminated Intravascular Coagulation in an Intensive Care Unit. American Journal of Hematology 2004, 76, 225–229.
- Levi, M.; Cate, H. ten Disseminated Intravascular Coagulation. The New England Journal of Medicine 1999, 341, 586–592, doi:10.1056/NEJM199908193410807.
- Gando, S.; Saitoh, D.; Ishikura, H.; Ueyama, M.; Otomo, Y.; Oda, S.; Kushimoto, S.; Tanjoh, K.; Mayumi, T.; Ikeda, T.; et al. A Randomized, Controlled, Multicenter Trial of the Effects of Antithrombin on Disseminated Intravascular Coagulation in Patients with Sepsis. Critical Care 2013, 17, doi:10.1186/cc13163.
- Masuda, T.; Shoko, T. Clinical Investigation of the Utility of a Pair of Coagulation-Fibrinolysis Markers for Definite Diagnosis of Sepsis-Induced Disseminated Intravascular Coagulation: A Single-Center, Diagnostic, Prospective, Observational Study. Thrombosis Research 2020, 192, 116–121, doi:10.1016/j.thromres.2020.05.009.
- Matsuda, K.; Kurokawa, M. Underlying Disease and Clinical Phenotypes of Disseminated Intravascular Coagulation. JMA Journal 2020, 3, 357–358, doi:10.31662/jmaj.2020-0072.
- Ohbe, H.; Yamakawa, K.; Taniguchi, K.; Morita, K.; Matsui, H.; Fushimi, K. Underlying Disorders, Clinical Phenotypes, and Treatment Diversity among Patients with Disseminated Intravascular Coagulation. JMA Journal 2020, 3, 321–329, doi:10.31662/jmaj.2020-0023.
- Jackson Chornenki, N.L.; Dwivedi, D.J.; Kwong, A.C.; Zamir, N.; Fox-Robichaud, A.E.; Liaw, P.C. Identification of Hemostatic Markers That Define the Pre-DIC State: A Multi-Center Observational Study. Journal of Thrombosis and Haemostasis 2020, 18, 2524–2531, doi:10.1111/jth.14973.
- Smith, L. Disseminated Intravascular Coagulation. Seminars in Oncology Nursing 2021, 000, 151135, doi:10.1016/j.soncn.2021.151135.
- Yamakawa, K.; Yoshimura, J.; Ito, T.; Hayakawa, M.; Hamasaki, T.; Fujimi, S. External Validation of the Two Newly Proposed Criteria for Assessing Coagulopathy in Sepsis. Thrombosis and Haemostasis 2019, 119, 203–212, doi:10.1055/s-0038-1676610.
- Naime, A.C.A.; Ganaes, J.O.F.; Lopes-Pires, M.E. Sepsis: The Involvement of Platelets and the Current Treatments. Current Molecular Pharmacology 2018, 11, 261–269, doi:10.2174/1874467211666180619124531.
- Masuda, T.; Shoko, T. Clinical Investigation of the Utility of a Pair of Coagulation-Fibrinolysis Markers for Definite Diagnosis of Sepsis-Induced Disseminated Intravascular Coagulation: A Single-Center, Diagnostic, Prospective, Observational Study. Thrombosis Research 2020, 192, 116–121, doi:10.1016/j.thromres.2020.05.009.
- Helms, J.; Severac, F.; Merdji, H.; Clere-Jehl, R.; François, B.; Mercier, E.; Quenot, J.P.; Meziani, F. Performances of Disseminated Intravascular Coagulation Scoring Systems in Septic Shock Patients. Annals of Intensive Care 2020, 10, doi:10.1186/s13613-020-00704-5.
- Abrams, S.T.; Morton, B.; Alhamdi, Y.; Alsabani, M.; Lane, S.; Welters, I.D.; Wang, G.; Toh, C.H. A Novel Assay for Neutrophil Extracellular Trap Formation Independently Predicts Disseminated Intravascular Coagulation and Mortality in Critically Ill Patients. American Journal of Respiratory and Critical Care Medicine 2019, 200, 869–880, doi:10.1164/rccm.201811-2111OC.
- Patel, P.; Walborn, A.; Rondina, M.; Fareed, J.; Hoppensteadt, D. Markers of Inflammation and Infection in Sepsis and Disseminated Intravascular Coagulation. Clinical and Applied Thrombosis/Hemostasis 2019, 25, doi:10.1177/1076029619843338.
- Yang, X.; Cheng, X.; Tang, Y.; Qiu, X.; Wang, Y.; Kang, H.; Wu, J.; Wang, Z.; Liu, Y.; Chen, F.; et al. Bacterial Endotoxin Activates the Coagulation Cascade through Gasdermin D-Dependent Phosphatidylserine Exposure. Immunity 2019, 51, 983-996.e6, doi:10.1016/j.immuni.2019.11.005.
- Kinasewitz, G.T.; Yan, S.B.; Basson, B.; Comp, P.; Russell, J.A.; Cariou, A.; Um, S.L.; Utterback, B.; Laterre, P.F.; Dhainaut, J.F. Universal Changes in Biomarkers of Coagulation and Inflammation Occur in Patients with Severe Sepsis, Regardless of Causative Micro-Organism [ISRCTN74215569]. Critical care (London, England) 2004, 8, 82–90, doi:10.1186/cc2459.
- Levi, M.; van der Poll, T. Coagulation and Sepsis. Thrombosis Research 2017, 149, 38–44, doi:10.1016/j.thromres.2016.11.007.
- Scully, M.; Levi, M. How We Manage Haemostasis during Sepsis. British Journal of Haematology 2019, 185, 209–218.
- Nieuwland, R.; Gardiner, C.; Dignat-George, F.; Mullier, F.; Mackman, N.; Woodhams, B.; Thaler, J. Toward Standardization of Assays Measuring Extracellular Vesicle-Associated Tissue Factor Activity. Journal of Thrombosis and Haemostasis 2019, 17, 1261–1264, doi:10.1111/jth.14481.
- Kay, J.G.; Grinstein, S. Phosphatidylserine-Mediated Cellular Signaling. Advances in Experimental Medicine and Biology 2013, 991, 177–193, doi:10.1007/978-94-007-6331-9_10.
- Delabranche, X.; Boisramé-Helms, J.; Asfar, P.; Berger, A.; Mootien, Y.; Lavigne, T.; Grunebaum, L.; Lanza, F.; Gachet, C.; Freyssinet, J.M.; et al. Microparticles Are New Biomarkers of Septic Shock-Induced Disseminated Intravascular Coagulopathy. Intensive Care Medicine 2013, 39, 1695–1703, doi:10.1007/s00134-013-2993-x.
- Matsumoto, H.; Yamakawa, K.; Ogura, H.; Koh, T.; Matsumoto, N.; Shimazu, T. Enhanced Expression of Cell-Specific Surface Antigens on Endothelial Microparticles in Sepsis-Induced Disseminated Intravascular Coagulation. Shock 2015, 43, 443–449, doi:10.1097/SHK.0000000000000331.
- Walborn, A.; Rondina, M.; Mosier, M.; Fareed, J.; Hoppensteadt, D. Endothelial Dysfunction Is Associated with Mortality and Severity of Coagulopathy in Patients with Sepsis and Disseminated Intravascular Coagulation. Clinical and Applied Thrombosis/Hemostasis 2019, 25, doi:10.1177/1076029619852163.
- VanTeeffelen, J.W.; Brands, J.; Stroes, E.S.; Vink, H. Endothelial Glycocalyx: Sweet Shield of Blood Vessels. Trends in Cardiovascular Medicine 2007, 17, 101–105, doi:10.1016/j.tcm.2007.02.002.
- Lupu, F.; Kinasewitz, G.; Dormer, K. The Role of Endothelial Shear Stress on Haemodynamics, Inflammation, Coagulation and Glycocalyx during Sepsis. Journal of Cellular and Molecular Medicine 2020, 24, 12258–12271.
- Sampei, S.; Okada, H.; Tomita, H.; Takada, C.; Suzuki, K.; Kinoshita, T.; Kobayashi, R.; Fukuda, H.; Kawasaki, Y.; Nishio, A.; et al. Endothelial Glycocalyx Disorders May Be Associated With Extended Inflammation During Endotoxemia in a Diabetic Mouse Model. Frontiers in Cell and Developmental Biology 2021, 9, doi:10.3389/fcell.2021.623582.
- Kushimoto, S.; Abe, T.; Ogura, H.; Shiraishi, A.; Saitoh, D.; Fujishima, S.; Mayumi, T.; Hifumi, T.; Shiino, Y.; Nakada, T. aki; et al. Impact of Blood Glucose Abnormalities on Outcomes and Disease Severity in Patients with Severe Sepsis: An Analysis from a Multicenter, Prospective Survey of Severe Sepsis. PLoS ONE 2020, 15, 1–15, doi:10.1371/journal.pone.0229919.
- Iba, T.; Connors, J.M.; Nagaoka, I.; Levy, J.H. Recent Advances in the Research and Management of Sepsis-Associated DIC. International Journal of Hematology 2021, 113, 24–33, doi:10.1007/s12185-020-03053-y.
- Hou, P.C.; Filbin, M.R.; Wang, H.; Ngo, L.; Aird, W.C.; Shapiro, N.I.; Wang, H.; Huang, D.T.; Angus, D.C.; Kellum, J.A.; et al. Endothelial Permeability and Hemostasis in Septic Shock: Results From the ProCESS Trial. Chest 2017, 152, 22–31, doi:10.1016/j.chest.2017.01.010.
- Portier, I.; Campbell, R.A. Role of Platelets in Detection and Regulation of Infection. Arteriosclerosis, Thrombosis, and Vascular Biology 2020, 70–78, doi:10.1161/ATVBAHA.120.314645.
- Ma, R.; Xie, R.; Yu, C.; Si, Y.; Wu, X.; Zhao, L.; Yao, Z.; Fang, S.; Chen, H.; Novakovic, V.; et al. Phosphatidylserine-Mediated Platelet Clearance by Endothelium Decreases Platelet Aggregates and Procoagulant Activity in Sepsis. Scientific Reports 2017, 7, 1–14, doi:10.1038/s41598-017-04773-8.
- Puskarich, M.A.; Cornelius, D.C.; Bandyopadhyay, S.; McCalmon, M.; Tramel, R.; Dale, W.D.; Jones, A.E. Phosphatidylserine Expressing Platelet Microparticle Levels at Hospital Presentation Are Decreased in Sepsis Non-Survivors and Correlate with Thrombocytopenia. Thrombosis Research 2018, 168, 138–144, doi:10.1016/j.thromres.2018.06.017.
- Tsirigotis, P.; Chondropoulos, S.; Frantzeskaki, F.; Stamouli, M.; Gkirkas, K.; Bartzeliotou, A.; Papanikolaou, N.; Atta, M.; Papassotiriou, I.; Dimitriadis, G.; et al. Thrombocytopenia in Critically Ill Patients with Severe Sepsis/Septic Shock: Prognostic Value and Association with a Distinct Serum Cytokine Profile. Journal of Critical Care 2016, 32, 9–15, doi:10.1016/j.jcrc.2015.11.010.
- Lopes-Pires, M.E.; Naime, A.C.A.; Cardelli, N.J.A.; Anjos, D.J.; Antunes, E.; Marcondes, S. PKC and AKT Modulate CGMP/PKG Signaling Pathway on Platelet Aggregation in Experimental Sepsis. PLoS ONE 2015, 10, 1–14, doi:10.1371/journal.pone.0137901.
- Naime, A.C.A.; Bonfitto, P.H.L.; Solon, C.; Lopes-Pires, M.E.; Anhê, G.F.; Antunes, E.; Marcondes, S. Tumor Necrosis Factor Alpha Has a Crucial Role in Increased Reactive Oxygen Species Production in Platelets of Mice Injected with Lipopolysaccharide. Platelets 2019, 30, 1047–1052, doi:10.1080/09537104.2019.1588241.
- Laursen, M.A.; Larsen, J.B.; Larsen, K.M.; Hvas, A.M. Platelet Function in Patients with Septic Shock. Thrombosis Research 2020, 185, 33–42, doi:10.1016/j.thromres.2019.11.011.
- Bardoel, B.W.; Kenny, E.F.; Sollberger, G.; Zychlinsky, A. The Balancing Act of Neutrophils. Cell Host and Microbe 2014, 15, 526–536, doi:10.1016/j.chom.2014.04.011.
- Metzler, K.D.; Goosmann, C.; Lubojemska, A.; Zychlinsky, A.; Papayannopoulos, V. Myeloperoxidase-Containing Complex Regulates Neutrophil Elastase Release and Actin Dynamics during NETosis. Cell Reports 2014, 8, 883–896, doi:10.1016/j.celrep.2014.06.044.
- Xie, T.; Duan, Z.; Sun, S.; Chu, C.; Ding, W. β-Lactams Modulate Neutrophil Extracellular Traps Formation Mediated by MTOR Signaling Pathway. Biochemical and Biophysical Research Communications 2021, 534, 408–414, doi:10.1016/j.bbrc.2020.11.067.
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.V.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-Induced NETosis Is a Dynamic Process Involving Neutrophil Multitasking in Vivo. Nature Medicine 2012, 18, 1386–1393, doi:10.1038/nm.2847.
- Shimizu, M.; Konishi, A.; Nomura, S. Examination of Biomarker Expressions in Sepsis-Related DIC Patients. International Journal of General Medicine 2018, 11, 353–361, doi:10.2147/IJGM.S173684.
- Jiao, Y.; Li, W.; Wang, W.; Tong, X.; Xia, R.; Fan, J.; Du, J.; Zhang, C.; Shi, X. Platelet-Derived Exosomes Promote Neutrophil Extracellular Trap Formation during Septic Shock. Critical Care 2020, 24, 1–18, doi:10.1186/s13054-020-03082-3.
- Rodrigues, D.A.S.; Prestes, E.B.; Gama, A.M.S.; de Souza Silva, L.; Pinheiro, A.A.S.; Ribeiro, J.M.C.; Campos, R.M.P.; Pimentel-Coelho, P.M.; de Souza, H.S.; Dicko, A.; et al. CXCR4 and MIF Are Required for Neutrophil Extracellular Trap Release Triggered by Plasmodium-Infected Erythrocytes. PLoS Pathogens 2020, 16, 1–23, doi:10.1371/JOURNAL.PPAT.1008230.
- Colón, D.F.; Wanderley, C.W.; Franchin, M.; Silva, C.M.; Hiroki, C.H.; Castanheira, F.V.S.; Donate, P.B.; Lopes, A.H.; Volpon, L.C.; Kavaguti, S.K.; et al. Neutrophil Extracellular Traps (NETs) Exacerbate Severity of Infant Sepsis. Critical Care 2019, 23, 1–13, doi:10.1186/s13054-019-2407-8.
- Huang, H.; Tohme, S.; Al-Khafaji, A.B.; Tai, S.; Loughran, P.; Chen, L.; Wang, S.; Kim, J.; Billiar, T.; Wang, Y.; et al. Damage-Associated Molecular Pattern-Activated Neutrophil Extracellular Trap Exacerbates Sterile Inflammatory Liver Injury. Hepatology 2015, 62, 600–614, doi:10.1002/hep.27841.
- Shrestha, B.; Ito, T.; Kakuuchi, M.; Totoki, T.; Nagasato, T.; Yamamoto, M.; Maruyama, I. Recombinant Thrombomodulin Suppresses Histone-Induced Neutrophil Extracellular Trap Formation. Frontiers in Immunology 2019, 10, 1–14, doi:10.3389/fimmu.2019.02535.
- Okeke, E.B.; Louttit, C.; Fry, C.; Najafabadi, A.H.; Han, K.; Nemzek, J.; Moon, J.J. Inhibition of Neutrophil Elastase Prevents Neutrophil Extracellular Trap Formation and Rescues Mice from Endotoxic Shock. Biomaterials 2020, 238, 119836, doi:10.1016/j.biomaterials.2020.119836.
- Kumar, S.; Gupta, E.; Kaushik, S.; Srivastava, V.K.; Saxena, J.; Mehta, S.; Jyoti, A. Quantification of NETs Formation in Neutrophil and Its Correlation with the Severity of Sepsis and Organ Dysfunction. Clinica Chimica Acta 2019, 495, 606–610, doi:10.1016/j.cca.2019.06.008.
- Chirivi, R.G.S.; van Rosmalen, J.W.G.; van der Linden, M.; Euler, M.; Schmets, G.; Bogatkevich, G.; Kambas, K.; Hahn, J.; Braster, Q.; Soehnlein, O.; et al. Therapeutic ACPA Inhibits NET Formation: A Potential Therapy for Neutrophil-Mediated Inflammatory Diseases. Cellular and Molecular Immunology 2020, 1–17, doi:10.1038/s41423-020-0381-3.
- Locke, M.; Francis, R.J.; Tsaousi, E.; Longstaff, C. Fibrinogen Protects Neutrophils from the Cytotoxic Effects of Histones and Delays Neutrophil Extracellular Trap Formation Induced by Ionomycin. Scientific Reports 2020, 10, 1–16, doi:10.1038/s41598-020-68584-0.
- Ode, Y.; Aziz, M.; Jin, H.; Arif, A.; Nicastro, J.G.; Wang, P. Cold-Inducible RNA-Binding Protein Induces Neutrophil Extracellular Traps in the Lungs during Sepsis. Scientific Reports 2019, 9, 1–11, doi:10.1038/s41598-019-42762-1.
- Levi, M.; Scully, M.; Singer, M. The Role of ADAMTS-13 in the Coagulopathy of Sepsis. Journal of Thrombosis and Haemostasis 2018, 16, 646–651.
- Hakkim, A.; Fürnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of Neutrophil Extracellular Trap Degradation Is Associated with Lupus Nephritis. Proceedings of the National Academy of Sciences of the United States of America 2010, 107, 9813–9818, doi:10.1073/pnas.0909927107.
- Müller, F.; Mutch, N.J.; Schenk, W.A.; Smith, S.A.; Esterl, L.; Spronk, H.M.; Schmidbauer, S.; Gahl, W.A.; Morrissey, J.H.; Renné, T. Platelet Polyphosphates Are Proinflammatory and Procoagulant Mediators In Vivo. Cell 2009, 139, 1143–1156, doi:10.1016/j.cell.2009.11.001.
- Smith, S.A.; Mutch, N.J.; Baskar, D.; Rohloff, P.; Docampo, R.; Morrissey, J.H. Polyphosphate Modulates Blood Coagulation and Fibrinolysis. Proceedings of the National Academy of Sciences 2006, 103, 903–908, doi:10.1073/pnas.0507195103.
- Finkelstein, E.; Rosen, G.M.; Rauckman, E.J. Spin Trapping of Superoxide and Hydroxyl Radical: Practical Aspects. Archives of Biochemistry and Biophysics 1980, 200, 1–16, doi:10.1016/0003-9861(80)90323-9.
- Griendling, K.K.; Touyz, R.M.; Zweier, J.L.; Dikalov, S.; Chilian, W.; Chen, Y.R.; Harrison, D.G.; Bhatnagar, A. Measurement of Reactive Oxygen Species, Reactive Nitrogen Species, and Redox-Dependent Signaling in the Cardiovascular System: A Scientific Statement from the American Heart Association; 2016; Vol. 119; ISBN 0000000000000.
- Kohen, R.; Nyska, A. Oxidation of Biological Systems: Oxidative Stress Phenomena, Antioxidants, Redox Reactions, and Methods for Their Quantification. Toxicologic Pathology 2002, 30, 620–650, doi:10.1080/01926230290166724.
- Nathan, C. Specificity of a Third Kind: Reactive Oxygen and Nitrogen Intermediates in Cell Signaling. Journal of Clinical Investigation 2003, 111, 769–778, doi:10.1172/JCI200318174.
- Nathan, C.; Xie, Q.W. Regulation of Biosynthesis of Nitric Oxide. Journal of Biological Chemistry 1994, 269, 13725–13728, doi:10.1016/s0021-9258(17)36703-0.
- Gkaliagkousi, E.; Ritter, J.; Ferro, A. Platelet-Derived Nitric Oxide Signaling and Regulation. Circulation Research 2007, 101.
- Doctor, A.; Platt, R.; Sheram, M.L.; Eischeid, A.; McMahon, T.; Maxey, T.; Doherty, J.; Axelrod, M.; Kline, J.; Gurka, M.; et al. Hemoglobin Conformation Couples Erythrocyte S-Nitrosothiol Content to O2 Gradients. Proceedings of the National Academy of Sciences of the United States of America 2005, 102, 5709–5714, doi:10.1073/pnas.0407490102.
- Jia, Y.; Xu, L.; Turner, D.J.; Martin, J.G. Endogenous Nitric Oxide Contributes to Strain-Related Differences in Airway Responsiveness in Rats. Journal of applied physiology (Bethesda, Md. : 1985) 1996, 80, 404–410, doi:10.1152/jappl.1996.80.2.404.
- Eiserich, J.P.; Hayakawa, K.A.; Cross, C.E. Sepsis and Hypotension: Enter Kynurenine, Move over Nitric Oxide. Critical Care Medicine 2011, 39.
- Titheradge, M.A. Nitric Oxide in Septic Shock. Biochimica et Biophysica Acta - Bioenergetics 1999, 1411.
- Winkler, M.S.; Kluge, S.; Holzmann, M.; Moritz, E.; Robbe, L.; Bauer, A.; Zahrte, C.; Priefler, M.; Schwedhelm, E.; Böger, R.H.; et al. Markers of Nitric Oxide Are Associated with Sepsis Severity: An Observational Study. Critical Care 2017, 21, doi:10.1186/s13054-017-1782-2.
- Lagan, A.L.; Melley, D.D.; Evans, T.W.; Quinlan, G.J. Pathogenesis of the Systemic Inflammatory Syndrome and Acute Lung Injury: Role of Iron Mobilization and Decompartmentalization. American Journal of Physiology - Lung Cellular and Molecular Physiology 2008, 294.
- Melley, D.D.; Finney, S.J.; Elia, A.; Lagan, A.L.; Quinlan, G.J.; Evans, T.W. Arterial Carboxyhemoglobin Level and Outcome in Critically Ill Patients. Critical Care Medicine 2007, 35, 1882–1887, doi:10.1097/01.CCM.0000275268.94404.43.
- Gutteridge, J.M.C.; Mumby, S.; Quinlan, G.J.; Chung, K.F.; Evans, T.W. Pro-Oxidant Iron Is Present in Human Pulmonary Epithelial Lining Fluid: Implications for Oxidative Stress in the Lung; 1996;
- Gutteridge, J.M.C.; Quinlan, G.J.; Evans, T.W. Transient Iron Overload with Bleomycin Detectable Iron in the Plasma of Patients with Adult Respiratory Distress Syndrome. Thorax 1994, 49, 707–710, doi:10.1136/thx.49.7.707.
- Gutteridge, J.M.C.; Quinlan, G.J. Antioxidant Protection against Organic and Inorganic Oxygen Radicals by Normal Human Plasma: The Important Primary Role for Iron-Binding and Iron-Oxidising Proteins; 1993;
- Cognasse, F.; Nguyen, K.A.; Damien, P.; McNicol, A.; Pozzetto, B.; Hamzeh-Cognasse, H.; Garraud, O. The Inflammatory Role of Platelets via Their TLRs and Siglec Receptors. Frontiers in Immunology 2015, 6.
- Cooper, D.; Stokes, K.Y.; Tailor, A.; Granger, D.N. Oxidative Stress Promotes Blood Cell-Endothelial Cell Interactions in the Microcirculation. Cardiovascular Toxicology 2002, 2.
- Joffre, J.; Hellman, J. Oxidative Stress and Endothelial Dysfunction in Sepsis and Acute Inflammation. Antioxidants & Redox Signaling 2021, doi:10.1089/ars.2021.0027.
- Krötz, F.; Sohn, H.Y.; Pohl, U. Reactive Oxygen Species: Players in the Platelet Game. Arteriosclerosis, Thrombosis, and Vascular Biology 2004, 24.
- Tyml, K. Critical Role for Oxidative Stress, Platelets, and Coagulation in Capillary Blood Flow Impairment in Sepsis. Microcirculation 2011, 18, 152–162, doi:10.1111/j.1549-8719.2010.00080.x.
- Dworakowski, R.; Alom-Ruiz, S.P.; Shah, A.M. NADPH Oxidase-Derived Reactive Oxygen Species in the Regulation of Endothelial Phenotype. In Proceedings of the Pharmacological Reports; 2008; Vol. 60.
- Koju, N.; Taleb, A.; Zhou, J.; Lv, G.; Yang, J.; Cao, X.; Lei, H.; Ding, Q. Pharmacological Strategies to Lower Crosstalk between Nicotinamide Adenine Dinucleotide Phosphate (NADPH) Oxidase and Mitochondria. Biomedicine and Pharmacotherapy 2019, 111.
- Magnani, F.; Nenci, S.; Fananas, E.M.; Ceccon, M.; Romero, E.; Fraaije, M.W.; Mattevi, A. Crystal Structures and Atomic Model of NADPH Oxidase. Proceedings of the National Academy of Sciences of the United States of America 2017, 114, doi:10.1073/pnas.1702293114.
- de Carvalho, D.D.; Sadok, A.; Bourgarel-Rey, V.; Gattacceca, F.; Penel, C.; Lehmann, M.; Kovacic, H. Nox1 Downstream of 12-Lipoxygenase Controls Cell Proliferation but Not Cell Spreading of Colon Cancer Cells. International Journal of Cancer 2008, 122, doi:10.1002/ijc.23300.
- Shin, S.W.; Seo, C.Y.; Han, H.; Han, J.Y.; Jeong, J.S.; Kwak, J.Y.; Park, J.I. 15d-PGJ2 Induces Apoptosis by Reactive Oxygen Species-Mediated Inactivation of Akt in Leukemia and Colorectal Cancer Cells and Shows in Vivo Antitumor Activity. Clinical Cancer Research 2009, 15, doi:10.1158/1078-0432.CCR-08-3101.
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free Radicals and Antioxidants in Normal Physiological Functions and Human Disease. The international journal of biochemistry & cell biology 2007, 39, 44–84, doi:10.1016/j.biocel.2006.07.001.
- Winterbourn, C.C. Toxicity of Iron and Hydrogen Peroxide: The Fenton Reaction. Toxicology Letters 1995, 82–83, doi:10.1016/0378-4274(95)03532-X.
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z. bin Mitochondrial Electron Transport Chain, ROS Generation and Uncoupling (Review). International Journal of Molecular Medicine 2019, 44.
- Cho, K.J.; Seo, J.M.; Kim, J.H. Bioactive Lipoxygenase Metabolites Stimulation of NADPH Oxidases and Reactive Oxygen Species. Molecules and Cells 2011, 32.
- Yun, M.R.; Park, H.M.; Seo, K.W.; Lee, S.J.; Im, D.S.; Kim, C.D. 5-Lipoxygenase Plays an Essential Role in 4-HNE-Enhanced ROS Production in Murine Macrophages via Activation of NADPH Oxidase. Free Radical Research 2010, 44, doi:10.3109/10715761003758122.
- Battelli, M.G.; Polito, L.; Bortolotti, M.; Bolognesi, A. Xanthine Oxidoreductase-Derived Reactive Species: Physiological and Pathological Effects. Oxidative Medicine and Cellular Longevity 2016, 2016.
- Nathan, C.; Xie, Q.W. Regulation of Biosynthesis of Nitric Oxide. Journal of Biological Chemistry 1994, 269.
- Marcondes, S.; Cardoso, M.H.M.; Morganti, R.P.; Thomazzi, S.M.; Lilla, S.; Murad, F.; de Nucci, G.; Antunes, E. Cyclic GMP-Independent Mechanisms Contribute to the Inhibition of Platelet Adhesion by Nitric Oxide Donor: A Role for-Actinin Nitration; 2006;
- Nieuwdorp, M.; Meuwese, M.C.; Mooij, H.L.; van Lieshout, M.H.P.; Hayden, A.; Levi, M.; Meijers, J.C.M.; Ince, C.; Kastelein, J.J.P.; Vink, H.; et al. Tumor Necrosis Factor-α Inhibition Protects against Endotoxin-Induced Endothelial Glycocalyx Perturbation. Atherosclerosis 2009, 202, doi:10.1016/j.atherosclerosis.2008.03.024.
- Schmidt, E.P.; Yang, Y.; Janssen, W.J.; Gandjeva, A.; Perez, M.J.; Barthel, L.; Zemans, R.L.; Bowman, J.C.; Koyanagi, D.E.; Yunt, Z.X.; et al. The Pulmonary Endothelial Glycocalyx Regulates Neutrophil Adhesion and Lung Injury during Experimental Sepsis. Nature Medicine 2012, 18, doi:10.1038/nm.2843.
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel Cell Death Program Leads to Neutrophil Extracellular Traps. Journal of Cell Biology 2007, 176, doi:10.1083/jcb.200606027.
- Manda-Handzlik, A.; Bystrzycka, W.; Cieloch, A.; Glodkowska-Mrowka, E.; Jankowska-Steifer, E.; Heropolitanska-Pliszka, E.; Skrobot, A.; Muchowicz, A.; Ciepiela, O.; Wachowska, M.; et al. Nitric Oxide and Peroxynitrite Trigger and Enhance Release of Neutrophil Extracellular Traps. Cellular and Molecular Life Sciences 2020, 77, doi:10.1007/s00018-019-03331-x.
- Grundmann, S.; Fink, K.; Rabadzhieva, L.; Bourgeois, N.; Schwab, T.; Moser, M.; Bode, C.; Busch, H.J. Perturbation of the Endothelial Glycocalyx in Post Cardiac Arrest Syndrome. Resuscitation 2012, 83, doi:10.1016/j.resuscitation.2012.01.028.
- Ince, C.; Mayeux, P.R.; Nguyen, T.; Gomez, H.; Kellum, J.A.; Ospina-Tascón, G.A.; Hernandez, G.; Murray, P.; de Backer, D. THE ENDOTHELIUM IN SEPSIS on Behalf of the ADQI XIV Workgroup. HHS public Access 2016, 45.
- Liu, Y.; Zhao, H.; Li, H.; Kalyanaraman, B.; Nicolosi, A.C.; Gutterman, D.D. Mitochondrial Sources of H2O2 Generation Play a Key Role in Flow-Mediated Dilation in Human Coronary Resistance Arteries. Circulation Research 2003, 93, doi:10.1161/01.RES.0000091261.19387.AE.
- Patil, N.K.; Parajuli, N.; Macmillan-Crow, L.A.; Mayeux, P.R. Inactivation of Renal Mitochondrial Respiratory Complexes and Manganese Superoxide Dismutase during Sepsis: Mitochondria-Targeted Antioxidant Mitigates Injury. American Journal of Physiology - Renal Physiology 2014, 306, doi:10.1152/ajprenal.00643.2013.
- Cimmino, G.; Cirillo, P.; Ragni, M.; Conte, S.; Uccello, G.; Golino, P. Reactive Oxygen Species Induce a Procoagulant State in Endothelial Cells by Inhibiting Tissue Factor Pathway Inhibitor. Journal of Thrombosis and Thrombolysis 2015, 40, doi:10.1007/s11239-015-1199-1.
- Altenhöfer, S.; Witte, I.; Teiber, J.F.; Wilgenbus, P.; Pautz, A.; Li, H.; Daiber, A.; Witan, H.; Clement, A.M.; Förstermann, U.; et al. One Enzyme, Two Functions: PON2 Prevents Mitochondrial Superoxide Formation and Apoptosis Independent from Its Lactonase Activity. Journal of Biological Chemistry 2010, 285, doi:10.1074/jbc.M110.118604.
- Ebert, J.; Wilgenbus, P.; Teiber, J.F.; Jurk, K.; Schwierczek, K.; Ohrmann, M.D.¨; Xia, N.; Li, H.; Spiecker, L.; Ruf, W.; et al. Paraoxonase-2 Regulates Coagulation Activation through Endothelial Tissue Factor; 2018;
- Wu, F.; Schuster, D.P.; Tyml, K.; Wilson, J.X. Ascorbate Inhibits NADPH Oxidase Subunit P47phox Expression in Microvascular Endothelial Cells. Free Radical Biology and Medicine 2007, 42, doi:10.1016/j.freeradbiomed.2006.10.033.
- Muzaffar, S.; Jeremy, J.Y.; Angelini, G.D.; Stuart-Smith, K.; Shukla, N. Role of the Endothelium and Nitric Oxide Synthases in Modulating Superoxide Formation Induced by Endotoxin and Cytokines in Porcine Pulmonary Arteries. Thorax 2003, 58, doi:10.1136/thorax.58.7.598.
- Cerwinka, W.H.; Cooper, D.; Krieglstein, C.F.; Ross, C.R.; McCord, J.M.; Granger, D.N. Superoxide Mediates Endotoxin-Induced Platelet-Endothelial Cell Adhesion in Intestinal Venules. American Journal of Physiology - Heart and Circulatory Physiology 2003, 284, doi:10.1152/ajpheart.00311.2002.
- Iba, T.; Levy, J.H. Derangement of the Endothelial Glycocalyx in Sepsis. Journal of Thrombosis and Haemostasis 2019, 17.
- Cheung, P.Y.; Salas, E.; Schulz, R.; Radomski, M.W. Nitric Oxide and Platelet Function: Implications for Neonatology. Seminars in Perinatology 1997, 21, doi:10.1016/S0146-0005(97)80006-7.
- Ulloa, L.; Cai, B.; Deitch, E.A. Novel Insights for Systemic Inflammation in Sepsis and Hemorrhage. Mediators of Inflammation 2010, 2010.
- Wang, L.; Taneja, R.; Razavi, H.M.; Law, C.; Gillis, C.; Mehta, S. Specific Role of Neutrophil Inducible Nitric Oxide Synthase in Murine Sepsis-Induced Lung Injury in Vivo. Shock 2012, 37, doi:10.1097/SHK.0b013e31824dcb5a.
- Farias Benjamim, C.; Santana Silva, J.; Bruno Fortes, Z.; Aparecida Oliveira, M.; Henrique Ferreira, S.; Queiroz Cunha, F. Inhibition of Leukocyte Rolling by Nitric Oxide during Sepsis Leads to Reduced Migration of Active Microbicidal Neutrophils. Infection and Immunity 2002, 70, doi:10.1128/IAI.70.7.3602-3610.2002.
- Iba, T.; Levy, J.H. Sepsis-Induced Coagulopathy and Disseminated Intravascular Coagulation. Anesthesiology 2020, 1, 1238–1245, doi:10.1097/ALN.0000000000003122.
- Lopes-Pires, M.E.; Clarke, S.R.; Marcondes, S.; Gibbins, J.M. Lipopolysaccharide Potentiates Platelet Responses via Toll-like Receptor 4-Stimulated Akt-Erk-PLA2 Signalling _ Enhanced Reader. Plos One 2017, 12.
- Masselli, E.; Pozzi, G.; Vaccarezza, M.; Mirandola, P.; Galli, D.; Vitale, M.; Carubbi, C.; Gobbi, G. ROS in Platelet Biology: Functional Aspects and Methodological Insights. International Journal of Molecular Sciences 2020, 21.
- Lopes-Pires, M.E.; Casarin, A.L.; Pereira-Cunha, F.G.; Lorand-Metze, I.; Antunes, E.; Marcondes, S. Lipopolysaccharide Treatment Reduces Rat Platelet Aggregation Independent of Intracellular Reactive-Oxygen Species Generation. Platelets 2012, 23, doi:10.3109/09537104.2011.603065.
- Dewitte, A.; Lepreux, S.; Villeneuve, J.; Rigothier, C.; Combe, C.; Ouattara, A.; Ripoche, J. Blood Platelets and Sepsis Pathophysiology: A New Therapeutic Prospect in Critical Ill Patients? Annals of Intensive Care 2017, 7.
- Zhang, G.; Zhang, P.; Liu, H.; Liu, X.; Xie, S.; Wang, X.; Wu, Y.; Chang, J.; Ma, L. Assessment of Th17/Treg Cells and Th Cytokines in an Improved Immune Thrombocytopenia Mouse Model. Hematology 2017, 22, 493–500, doi:10.1080/10245332.2017.1301040.
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Frontiers in Cellular and Infection Microbiology 2017, 7.
- Fuentes, E.; Gibbins, J.M.; Holbrook, L.M.; Palomo, I. NADPH Oxidase 2 (NOX2): A Key Target of Oxidative Stress-Mediated Platelet Activation and Thrombosis. Trends in Cardiovascular Medicine 2018, 28.
- Seno, T.; Inoue, N.; Gao, D.; Okuda, M.; Sumi, Y.; Matsui, K.; Yamada, S.; Hirata, K.I.; Kawashima, S.; Tawa, R.; et al. Involvement of NADH/NADPH Oxidase in Human Platelet ROS Production. Thrombosis Research 2001, 103, doi:10.1016/S0049-3848(01)00341-3.
- Sonkar, V.K.; Kumar, R.; Jensen, M.; Wagner, B.A.; Sharathkumar, A.A.; Miller, F.J.; Fasano, M.B.; Lentz, S.R.; Buettner, G.R.; Dayal, S. Nox2 NADPH Oxidase Is Dispensable for Platelet Activation or Arterial Thrombosis in Mice. Blood Advances 2019, 3, doi:10.1182/bloodadvances.2018025569.
- Vara, D.; Tarafdar, A.; Celikag, M.; Patinha, D.; Gulacsy, C.E.; Hounslea, E.; Warren, Z.; Ferreira, B.; Koeners, M.P.; Caggiano, L.; et al. NADPH Oxidase 1 Is a Novel Pharmacological Target for the Development of an Antiplatelet Drug without Bleeding Side Effects. FASEB Journal 2020, 34, doi:10.1096/fj.202001086RRR.
- Lopes Pires, M.E.; Antunes Naime, A.C.; Oliveira, J.G.F.; Anhe, G.F.; Garraud, O.; Cognasse, F.; Antunes, E.; Marcondes, S. Signalling Pathways Involved in P47 Phox -Dependent Reactive Oxygen Species in Platelets of Endotoxemic Rats. Basic and Clinical Pharmacology and Toxicology 2019, 124, doi:10.1111/bcpt.13148.
- Naime, A.C.A.; Ganaes, J.O.F.; Lopes-Pires, M.E. Sepsis: The Involvement of Platelets and the Current Treatments. Current Molecular Pharmacology 2018, 11, 261–269, doi:10.2174/1874467211666180619124531.
- Clark, S.R.; Ma, A.C.; Tavener, S. a; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 Activates Neutrophil Extracellular Traps to Ensnare Bacteria in Septic Blood. Nature medicine 2007, 13, 463–469, doi:10.1038/nm1565.
- Parker, H.; Dragunow, M.; Hampton, M.B.; Kettle, A.J.; Winterbourn, C.C. Requirements for NADPH Oxidase and Myeloperoxidase in Neutrophil Extracellular Trap Formation Differ Depending on the Stimulus. Journal of Leukocyte Biology 2012, 92, doi:10.1189/jlb.1211601.
- Camicia, G.; Pozner, R.; de Larrañaga, G. Neutrophil Extracellular Traps in Sepsis. Shock 2014, 42, 286–294.
- Puskarich, M.A.; Kline, J.A.; Watts, J.A.; Shirey, K.; Hosler, J.; Jones, A.E. Early Alterations in Platelet Mitochondrial Function Are Associated with Survival and Organ Failure in Patients with Septic Shock. Journal of Critical Care 2016, 31, doi:10.1016/j.jcrc.2015.10.005.
- Sjövall, F.; Morota, S.; Hansson, M.J.; Friberg, H.; Gnaiger, E.; Elmér, E. Temporal Increase of Platelet Mitochondrial Respiration Is Negatively Associated with Clinical Outcome in Patients with Sepsis. Critical Care 2010, 14, doi:10.1186/cc9337.
- Mantzarlis, K.; Tsolaki, V.; Zakynthinos, E. Role of Oxidative Stress and Mitochondrial Dysfunction in Sepsis and Potential Therapies. Oxidative Medicine and Cellular Longevity 2017, 2017.
- Matsumoto, S.; Koga, H.; Kusaka, J.; Hagiwara, S. Effects of the Antioxidant-Enriched Concentrated Liquid Diet ANOM on Oxidative Stress and Multiple Organ Injury in Patients with Septic Shock: A Pilot Study. Journal of Anesthesia & Clinical Research 2011, 02, doi:10.4172/2155-6148.1000155.
- Shen, K.-P.; Lo, Y.-C.; Yang, R.-C.; Liu, H.-W.; Chen, I.-J.; Wu, B.-N. Antioxidant Eugenosedin-A Protects against Lipopolysaccharide-Induced Hypotension, Hyperglycaemia and Cytokine Immunoreactivity in Rats and Mice. Journal of Pharmacy and Pharmacology 2010, 57, 117–125, doi:10.1211/0022357055137.
- Pleiner, J.; Mittermayer, F.; Schaller, G.; Marsik, C.; Macallister, R.J.; Wolzt, M. Inflammation-Induced Vasoconstrictor Hyporeactivity Is Caused by Oxidative Stress. 2003, doi:10.1016/S0735-1097(03)01076-3.
- Sakr, Y.; Vincent, J.-L.; Schuerholz, T.; Filipescu, D.; Romain, A.; Hjelmqvist, H.; Reinhart, K. Early- versus Late-Onset Shock in European Intensive Care Units. Shock (Augusta, Ga.) 2007, 28, 636–643.
- Forceville, X.; Vitoux, D.; Gauzit, R.; Combes, A.; Lahilaire, P.; Chappuis, P. Selenium, Systemic Immune Response Syndrome, Sepsis, and Outcome in Critically Ill Patients. Critical Care Medicine 1998, 26, 1536–1544, doi:10.1097/00003246-199809000-00021.
- Angstwurm, M.W.A.; Engelmann, L.; Zimmermann, T.; Lehmann, C.; Spes, C.H.; Abel, P.; Strauß, R.; Meier-Hellmann, A.; Insel, R.; Radke, J.; et al. Selenium in Intensive Care (SIC): Results of a Prospective Randomized, Placebo-Controlled, Multiple-Center Study in Patients with Severe Systemic Inflammatory Response Syndrome, Sepsis, and Septic Shock. Critical Care Medicine 2007, 35, 118–126, doi:10.1097/01.CCM.0000251124.83436.0E.
- Manzanares, W.; Lemieux, M.; Elke, G.; Langlois, P.L.; Bloos, F.; Heyland, D.K. High-Dose Intravenous Selenium Does Not Improve Clinical Outcomes in the Critically Ill: A Systematic Review and Meta-Analysis. Critical care (London, England) 2016, 20, 356, doi:10.1186/s13054-016-1529-5.
- Pei, Y.; Liu, H.; Yang, Y.; Yang, Y.; Jiao, Y.; Tay, F.R.; Chen, J. Biological Activities and Potential Oral Applications of N-Acetylcysteine: Progress and Prospects. Oxidative Medicine and Cellular Longevity 2018, 2018.
- de Mello, R.O.; Lunardelli, A.; Caberlon, E.; de Moraes, C.M.B.; Christ Vianna Santos, R.; da Costa, V.L.; da Silva, G.V.; da Silva Scherer, P.; Buaes, L.E.C.; da Silva Melo, D.A.; et al. Effect of N-Acetylcysteine and Fructose-1,6-Bisphosphate in the Treatment of Experimental Sepsis. Inflammation 2011, 34, 539–550, doi:10.1007/s10753-010-9261-9.
- Ritter, C.; Reinke, A.; Andrades, M.; Martins, M.R.; Rocha, J.; Menna-Barreto, S.; Quevedo, J.; Moreira, J.C.F.; Dal-Pizzol, F. Protective Effect of N-Acetylcysteine and Deferoxamine on Carbon Tetrachloride-Induced Acute Hepatic Failure in Rats. Critical care medicine 2004, 32, 2079–2083, doi:10.1097/01.ccm.0000142699.54266.d9.
- Villa, P.; Saccani, A.; Sica, A.; Ghezzi, P. Glutathione Protects Mice from Lethal Sepsis by Limiting Inflammation and Potentiating Host Defense. The Journal of infectious diseases 2002, 185, 1115–1120, doi:10.1086/340042.
- Schmidt, W.; Walther, A.; Gebhard, M.M.; Martin, E.; Schmidt, H. Influence of N-Acetylcysteine Treatment on Endotoxin-Induced Microcirculatory Disturbances. Intensive care medicine 1998, 24, 967–972, doi:10.1007/s001340050697.
- Chertoff, J. N-Acetylcysteine’s Role in Sepsis and Potential Benefit in Patients With Microcirculatory Derangements. Journal of intensive care medicine 2018, 33, 87–96, doi:10.1177/0885066617696850.
- Ortolani, O.; Conti, A.; de Gaudio, A.R.; Moraldi, E.; Cantini, Q.; Novelli, G. The Effect of Glutathione and N-Acetylcysteine on Lipoperoxidative Damage in Patients with Early Septic Shock. American journal of respiratory and critical care medicine 2000, 161, 1907–1911, doi:10.1164/ajrccm.161.6.9903043.
- Spies, C.D.; Reinhart, K.; Witt, I.; Meier-Hellmann, A.; Hannemann, L.; Bredle, D.L.; Schaffartzik, W. Influence of N-Acetylcysteine on Indirect Indicators of Tissue Oxygenation in Septic Shock Patients: Results from a Prospective, Randomized, Double-Blind Study. Critical care medicine 1994, 22, 1738–1746.
- Bastin, A.J.; Davies, N.; Lim, E.; Quinlan, G.J.; Griffiths, M.J. Systemic Inflammation and Oxidative Stress Post-Lung Resection: Effect of Pretreatment with N-Acetylcysteine. Respirology (Carlton, Vic.) 2016, 21, 180–187, doi:10.1111/resp.12662.
- Heller, A.R.; Groth, G.; Heller, S.C.; Breitkreutz, R.; Nebe, T.; Quintel, M.; Koch, T. N-Acetylcysteine Reduces Respiratory Burst but Augments Neutrophil Phagocytosis in Intensive Care Unit Patients. Critical care medicine 2001, 29, 272–276, doi:10.1097/00003246-200102000-00009.
- Spapen, H.D. Effects of N-Acetylcysteine on Microalbuminuria and Organ Failure in Acute Severe Sepsis *. CHEST Journal 2005, 127, 1413, doi:10.1378/chest.127.4.1413.
- Rodriguez-Cuenca, S.; Cochemé, H.M.; Logan, A.; Abakumova, I.; Prime, T.A.; Rose, C.; Vidal-Puig, A.; Smith, A.C.; Rubinsztein, D.C.; Fearnley, I.M.; et al. Consequences of Long-Term Oral Administration of the Mitochondria-Targeted Antioxidant MitoQ to Wild-Type Mice. Free radical biology & medicine 2010, 48, 161–172, doi:10.1016/j.freeradbiomed.2009.10.039.
- Lowes, D.A.; Webster, N.R.; Murphy, M.P.; Galley, H.F. Antioxidants That Protect Mitochondria Reduce Interleukin-6 and Oxidative Stress, Improve Mitochondrial Function, and Reduce Biochemical Markers of Organ Dysfunction in a Rat Model of Acute Sepsis. British journal of anaesthesia 2013, 110, 472–480, doi:10.1093/bja/aes577.
- Okado-Matsumoto, A.; Fridovich, I. Subcellular Distribution of Superoxide Dismutases (SOD) in Rat Liver. Journal of Biological Chemistry 2001, 276, 38388–38393, doi:10.1074/jbc.M105395200.
- McCord, J.M.; Fridovich, I. Superoxide Dismutase. An Enzymic Function for Erythrocuprein (Hemocuprein). The Journal of biological chemistry 1969, 244, 6049–6055.
- Macarthur, H.; Couri, D.M.; Wilken, G.H.; Westfall, T.C.; Lechner, A.J.; Matuschak, G.M.; Chen, Z.; Salvemini, D. Modulation of Serum Cytokine Levels by a Novel Superoxide Dismutase Mimetic, M40401, in an Escherichia Coli Model of Septic Shock: Correlation with Preserved Circulating Catecholamines. Critical care medicine 2003, 31, 237–245, doi:10.1097/00003246-200301000-00037.
- Privalle, C.; Talarico, T.; Keng, T.; DeAngelo, J. Pyridoxalated Hemoglobin Polyoxyethylene: A Nitric Oxide Scavenger with Antioxidant Activity for the Treatment of Nitric Oxide-Induced Shock. Free radical biology & medicine 2000, 28, 1507–1517, doi:10.1016/s0891-5849(00)00260-4.
- Bone, H.G.; Schenarts, P.J.; Fischer, S.R.; McGuire, R.; Traber, L.D.; Traber, D.L. Pyridoxalated Hemoglobin Polyoxyethylene Conjugate Reverses Hyperdynamic Circulation in Septic Sheep. Journal of Applied Physiology 1998, 84, 1991–1999, doi:10.1152/jappl.1998.84.6.1991.
- Bone, H.G.; Schenarts, P.J.; Booke, M.; McGuire, R.; Harper, D.; Traber, L.D.; Traber, D.L. Oxalated Pyridoxalated Hemoglobin Polyoxyethylene Conjugate Normalizes the Hyperdynamic Circulation in Septic Sheep. Critical Care Medicine 1997, 25, 1010–1018, doi:10.1097/00003246-199706000-00019.
- Grover, R.; Zaccardelli, D.; Colice, G.; Guntupalli, K.; Watson, D.; Vincent, J.L. An Open-Label Dose Escalation Study of the Nitric Oxide Synthase Inhibitor, N(G)-Methyl-L-Arginine Hydrochloride (546C88), in Patients with Septic Shock. Glaxo Wellcome International Septic Shock Study Group. Critical care medicine 1999, 27, 913–922, doi:10.1097/00003246-199905000-00025.
- Vincent, J.-L.; Privalle, C.T.; Singer, M.; Lorente, J.A.; Boehm, E.; Meier-Hellmann, A.; Darius, H.; Ferrer, R.; Sirvent, J.-M.; Marx, G.; et al. Multicenter, Randomized, Placebo-Controlled Phase III Study of Pyridoxalated Hemoglobin Polyoxyethylene in Distributive Shock (PHOENIX). Critical care medicine 2015, 43, 57–64, doi:10.1097/CCM.0000000000000554.
- Loren, P.; Sánchez, R.; Arias, M.-E.; Felmer, R.; Risopatrón, J.; Cheuquemán, C. Melatonin Scavenger Properties against Oxidative and Nitrosative Stress: Impact on Gamete Handling and In Vitro Embryo Production in Humans and Other Mammals. International journal of molecular sciences 2017, 18, doi:10.3390/ijms18061119.
- Sener, G.; Toklu, H.; Kapucu, C.; Ercan, F.; Erkanli, G.; Kaçmaz, A.; Tilki, M.; Yeğen, B.C. Melatonin Protects against Oxidative Organ Injury in a Rat Model of Sepsis. Surgery today 2005, 35, 52–59, doi:10.1007/s00595-004-2879-1.
- Alamili, M.; Bendtzen, K.; Lykkesfeldt, J.; Rosenberg, J.; Gögenur, I. Melatonin Suppresses Markers of Inflammation and Oxidative Damage in a Human Daytime Endotoxemia Model. Journal of critical care 2014, 29, 184.e9-184.e13, doi:10.1016/j.jcrc.2013.09.006.
- Mantzarlis, K.; Tsolaki, V.; Zakynthinos, E. Role of Oxidative Stress and Mitochondrial Dysfunction in Sepsis and Potential Therapies. Oxidative medicine and cellular longevity 2017, 2017, 5985209, doi:10.1155/2017/5985209.