 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Isabella Russo | + 1944 word(s) | 1944 | 2021-12-30 07:34:31 | | | |
| 2 | Vicky Zhou | Meta information modification | 1944 | 2021-12-30 09:36:43 | | |
Video Upload Options
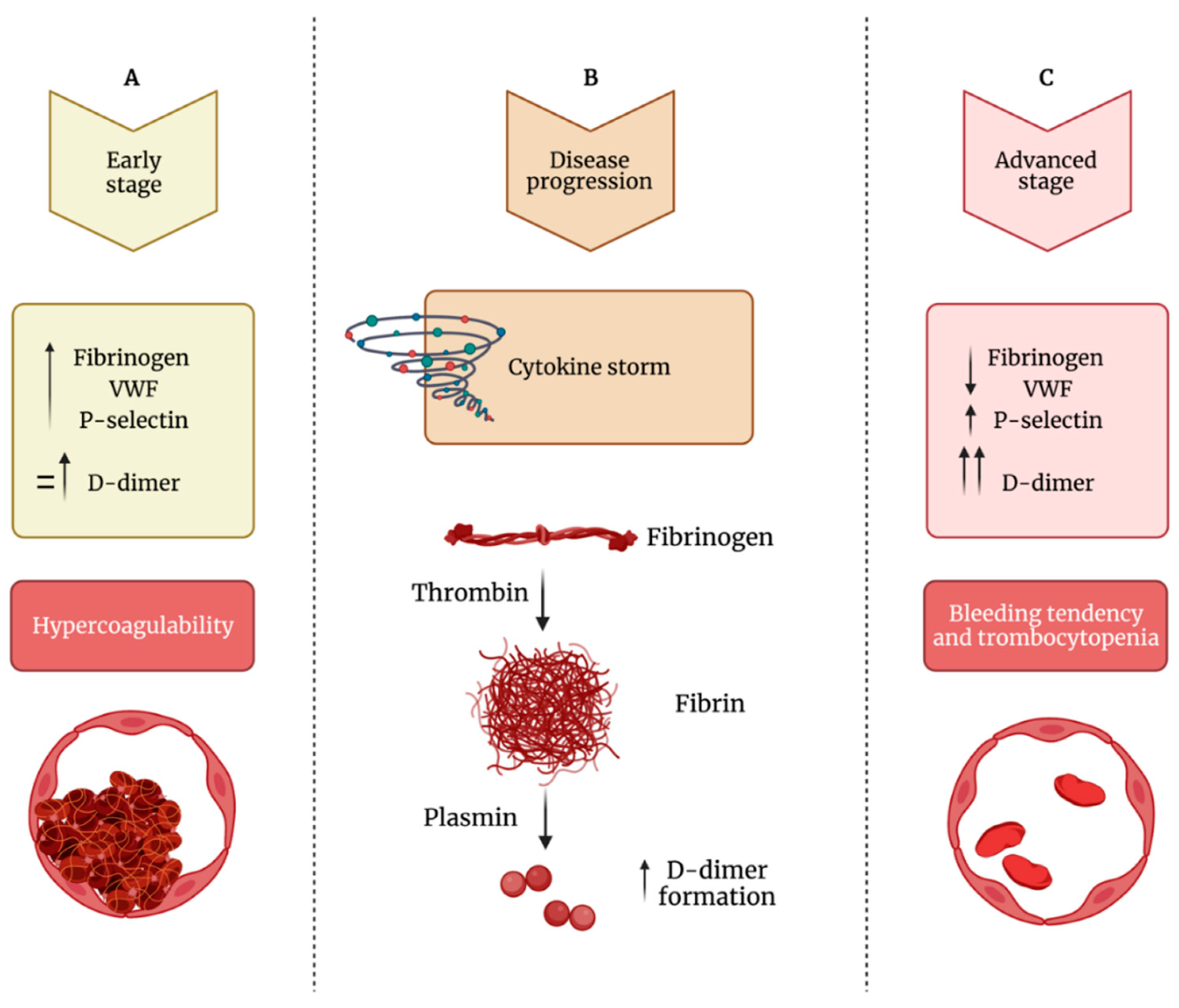
COVID-19 infection is associated with a broad spectrum of presentations, but alveolar capillary microthrombi have been described as a common finding in COVID-19 patients, appearing as a consequence of a severe endothelial injury with endothelial cell membrane disruption. These observations clearly point to the identification of a COVID-19-associated coagulopathy, which may contribute to thrombosis, multi-organ damage, and cause of severity and fatality. One significant finding that emerges in prothrombotic abnormalities observed in COVID-19 patients is that the coagulation alterations are mainly mediated by the activation of platelets and intrinsically related to viral-mediated endothelial inflammation. Beyond the well-known role in hemostasis, the ability of platelets to also release various potent cytokines and chemokines has elevated these small cells from simple cell fragments to crucial modulators in the blood, including their inflammatory functions, that have a large influence on the immune response during infectious disease. Indeed, platelets are involved in the pathogenesis of acute lung injury also by promoting NET formation and affecting vascular permeability. Specifically, the deposition by activated platelets of the chemokine platelet factor 4 at sites of inflammation promotes adhesion of neutrophils on endothelial cells and thrombogenesis, and it seems deeply involved in the phenomenon of vaccine-induced thrombocytopenia and thrombosis. Importantly, the hyperactivated platelet phenotype along with evidence of cytokine storm, high levels of P-selectin, D-dimer, and, on the other hand, decreased levels of fibrinogen, von Willebrand factor, and thrombocytopenia may be considered suitable biomarkers that distinguish the late stage of COVID-19 progression in critically ill patients.
1. Introduction
2. SARS-CoV-2 Effects on Platelets
3. Conclusions
References
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128.
- Yeaman, M.R. Bacterial-Platelet Interactions: Virulence Meets Host Defense. Future Microbiol. 2010, 5, 471–506.
- De Stoppelaar, S.F.; van’t Veer, C.; van der Poll, T. The Role of Platelets in Sepsis. Thromb. Haemost. 2014, 112, 666–677.
- Jenne, C.N.; Kubes, P. Platelets in Inflammation and Infection. Platelets 2015, 26, 286–292.
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basílio, J.; Petzelbauer, P.; Assinger, A.; et al. Cell Type-Specific Roles of NF-ΚB Linking Inflammation and Thrombosis. Front. Immunol. 2019, 10, 85.
- Liverani, E.; Mondrinos, M.J.; Sun, S.; Kunapuli, S.P.; Kilpatrick, L.E. Role of Protein Kinase C-Delta in Regulating Platelet Activation and Platelet-Leukocyte Interaction during Sepsis. PLoS ONE 2018, 13, e0195379.
- Acanfora, D.; Acanfora, C.; Ciccone, M.M.; Scicchitano, P.; Bortone, A.S.; Uguccioni, M.; Casucci, G. The Cross-Talk between Thrombosis and Inflammatory Storm in Acute and Long-COVID-19: Therapeutic Targets and Clinical Cases. Viruses 2021, 13, 1904.
- Cicala, C.; Cirino, G. Linkage between Inflammation and Coagulation: An Update on the Molecular Basis of the Crosstalk. Life Sci. 1998, 62, 1817–1824.
- Esmon, C.T.; Xu, J.; Lupu, F. Innate Immunity and Coagulation. J. Thromb. Haemost. 2011, 9 (Suppl. 1), 182–188.
- Taus, F.; Salvagno, G.; Canè, S.; Fava, C.; Mazzaferri, F.; Carrara, E.; Petrova, V.; Barouni, R.M.; Dima, F.; Dalbeni, A.; et al. Platelets Promote Thromboinflammation in SARS-CoV-2 Pneumonia. Arter. Thromb. Vasc. Biol. 2020, 40, 2975–2989.
- Hansson, G.K.; Robertson, A.-K.L.; Söderberg-Nauclér, C. Inflammation and Atherosclerosis. Annu. Rev. Pathol. 2006, 1, 297–329.
- Stark, K.; Massberg, S. Interplay between Inflammation and Thrombosis in Cardiovascular Pathology. Nat. Rev. Cardiol. 2021, 18, 666–682.
- McFadyen, J.D.; Stevens, H.; Peter, K. The Emerging Threat of (Micro)Thrombosis in COVID-19 and Its Therapeutic Implications. Circ. Res. 2020, 127, 571–587.
- Melillo, F.; Napolano, A.; Loffi, M.; Regazzoni, V.; Boccellino, A.; Danzi, G.B.; Cappelletti, A.M.; Rovere-Querini, P.; Landoni, G.; Ingallina, G.; et al. Myocardial Injury in Patients with SARS-CoV-2 Pneumonia: Pivotal Role of Inflammation in COVID-19. Eur. J. Clin. Investig. 2021, e13703.
- Attiq, A.; Yao, L.J.; Afzal, S.; Khan, M.A. The Triumvirate of NF-ΚB, Inflammation and Cytokine Storm in COVID-19. Int. Immunopharmacol. 2021, 101, 108255.
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet Gene Expression and Function in Patients with COVID-19. Blood 2020, 136, 1317–1329.
- Barrett, T.J.; Bilaloglu, S.; Cornwell, M.; Burgess, H.M.; Virginio, V.W.; Drenkova, K.; Ibrahim, H.; Yuriditsky, E.; Aphinyanaphongs, Y.; Lifshitz, M.; et al. Platelets Contribute to Disease Severity in COVID-19. J. Thromb. Haemost. 2021.
- Wang, W.; Xu, Y.; Gao, R.; Lu, R.; Han, K.; Wu, G.; Tan, W. Detection of SARS-CoV-2 in Different Types of Clinical Specimens. JAMA 2020, 323, 1843–1844.
- Campbell, R.A.; Boilard, E.; Rondina, M.T. Is There a Role for the ACE2 Receptor in SARS-CoV-2 Interactions with Platelets? J. Thromb. Haemost. 2021, 19, 46–50.
- Zhang, S.; Liu, Y.; Wang, X.; Yang, L.; Li, H.; Wang, Y.; Liu, M.; Zhao, X.; Xie, Y.; Yang, Y.; et al. SARS-CoV-2 Binds Platelet ACE2 to Enhance Thrombosis in COVID-19. J. Hematol. Oncol. 2020, 13, 120.
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical Features of Patients Infected with 2019 Novel Coronavirus in Wuhan, China. Lancet 2020, 395, 497–506.
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circ. Res. 2018, 122, 337–351.
- Koupenova, M.; Freedman, J.E. Platelets: The Unsung Hero of the Immune Response. J. Thromb. Haemost. 2015, 13, 268–270.
- Ding, Y.; Wang, H.; Shen, H.; Li, Z.; Geng, J.; Han, H.; Cai, J.; Li, X.; Kang, W.; Weng, D.; et al. The Clinical Pathology of Severe Acute Respiratory Syndrome (SARS): A Report from China. J. Pathol. 2003, 200, 282–289.
- Malha, L.; Mueller, F.B.; Pecker, M.S.; Mann, S.J.; August, P.; Feig, P.U. COVID-19 and the Renin-Angiotensin System. Kidney Int. Rep. 2020, 5, 563–565.
- Harrison, D.G.; Guzik, T.J.; Lob, H.E.; Madhur, M.S.; Marvar, P.J.; Thabet, S.R.; Vinh, A.; Weyand, C.M. Inflammation, Immunity, and Hypertension. Hypertension 2011, 57, 132–140.
- Dmitrieva, N.I.; Burg, M.B. Elevated Sodium and Dehydration Stimulate Inflammatory Signaling in Endothelial Cells and Promote Atherosclerosis. PLoS ONE 2015, 10, e0128870.
- Nemerson, Y. Tissue Factor and Hemostasis. Blood 1988, 71, 1–8.
- Mackman, N.; Tilley, R.E.; Key, N.S. Role of the Extrinsic Pathway of Blood Coagulation in Hemostasis and Thrombosis. Arter. Thromb. Vasc. Biol. 2007, 27, 1687–1693.
- Grover, S.P.; Mackman, N. Tissue Factor: An Essential Mediator of Hemostasis and Trigger of Thrombosis. Arter. Thromb. Vasc. Biol. 2018, 38, 709–725.
- Mackman, N. Regulation of the Tissue Factor Gene. Thromb. Haemost. 1997, 78, 747–754.
- Wang, J.; Mahmud, S.A.; Bitterman, P.B.; Huo, Y.; Slungaard, A. Histone Deacetylase Inhibitors Suppress TF-KappaB-Dependent Agonist-Driven Tissue Factor Expression in Endothelial Cells and Monocytes. J. Biol. Chem. 2007, 282, 28408–28418.
- Li, Y.-D.; Ye, B.-Q.; Zheng, S.-X.; Wang, J.-T.; Wang, J.-G.; Chen, M.; Liu, J.-G.; Pei, X.-H.; Wang, L.-J.; Lin, Z.-X.; et al. NF-KappaB Transcription Factor P50 Critically Regulates Tissue Factor in Deep Vein Thrombosis. J. Biol. Chem. 2009, 284, 4473–4483.
- Mackman, N. The Many Faces of Tissue Factor. J. Thromb. Haemost. 2009, 7 (Suppl. 1), 136–139.
- Rosell, A.; Havervall, S.; von Meijenfeldt, F.; Hisada, Y.; Aguilera, K.; Grover, S.P.; Lisman, T.; Mackman, N.; Thålin, C. Patients With COVID-19 Have Elevated Levels of Circulating Extracellular Vesicle Tissue Factor Activity That Is Associated with Severity and Mortality-Brief Report. Arter. Thromb. Vasc. Biol. 2021, 41, 878–882.
- Subrahmanian, S.; Borczuk, A.; Salvatore, S.; Fung, K.-M.; Merrill, J.T.; Laurence, J.; Ahamed, J. Tissue Factor Upregulation Is Associated with SARS-CoV-2 in the Lungs of COVID-19 Patients. J. Thromb. Haemost. 2021, 19, 2268–2274.
- Bury, L.; Camilloni, B.; Castronari, R.; Piselli, E.; Malvestiti, M.; Borghi, M.; KuchiBotla, H.; Falcinelli, E.; Petito, E.; Amato, F.; et al. Search for SARS-CoV-2 RNA in Platelets from COVID-19 Patients. Platelets 2021, 32, 284–287.
- Zaid, Y.; Puhm, F.; Allaeys, I.; Naya, A.; Oudghiri, M.; Khalki, L.; Limami, Y.; Zaid, N.; Sadki, K.; Ben, E.l.; et al. Platelets Can Associate with SARS-Cov-2 RNA and Are Hyperactivated in COVID-19. Circ. Res. 2020, 127, 1404–1418.
- Barrett, T.J.; Lee, A.H.; Xia, Y.; Lin, L.H.; Black, M.; Cotzia, P.; Hochman, J.; Berger, J.S. Platelet and Vascular Biomarkers Associate with Thrombosis and Death in Coronavirus Disease. Circ. Res. 2020, 127, 945–947.
- Shen, B.; Yi, X.; Sun, Y.; Bi, X.; Du, J.; Zhang, C.; Quan, S.; Zhang, F.; Sun, R.; Qian, L.; et al. Proteomic and Metabolomic Characterization of COVID-19 Patient Sera. Cell 2020, 182, 59–72.e15.
- Bergmann, C.C.; Silverman, R.H. COVID-19: Coronavirus Replication, Pathogenesis, and Therapeutic Strategies. Clevel. Clin. J. Med. 2020, 87, 321–327.
- Grobbelaar, L.M.; Venter, C.; Vlok, M.; Ngoepe, M.; Laubscher, G.J.; Lourens, P.J.; Steenkamp, J.; Kell, D.B.; Pretorius, E. SARS-CoV-2 Spike Protein S1 Induces Fibrin(Ogen) Resistant to Fibrinolysis: Implications for Microclot Formation in COVID-19. Biosci. Rep. 2021, 41, BSR20210611.
- Kawase, M.; Kataoka, M.; Shirato, K.; Matsuyama, S. Biochemical Analysis of Coronavirus Spike Glycoprotein Conformational Intermediates during Membrane Fusion. J. Virol. 2019, 93, e00785-19.
- Watanabe, Y.; Allen, J.D.; Wrapp, D.; McLellan, J.S.; Crispin, M. Site-Specific Glycan Analysis of the SARS-CoV-2 Spike. Science 2020, 369, 330–333.
- Flores-Alanis, A.; Sandner-Miranda, L.; Delgado, G.; Cravioto, A.; Morales-Espinosa, R. The Receptor Binding Domain of SARS-CoV-2 Spike Protein Is the Result of an Ancestral Recombination between the Bat-CoV RaTG13 and the Pangolin-CoV MP789. BMC Res. Notes 2020, 13, 398.
- Letarov, A.V.; Babenko, V.V.; Kulikov, E.E. Free SARS-CoV-2 Spike Protein S1 Particles May Play a Role in the Pathogenesis of COVID-19 Infection. Biochemistry 2021, 86, 257–261.
- Engelmann, B.; Massberg, S. Thrombosis as an Intravascular Effector of Innate Immunity. Nat. Rev. Immunol. 2013, 13, 34–45.
- Weyrich, A.S.; Zimmerman, G.A. Platelets in Lung Biology. Annu. Rev. Physiol. 2013, 75, 569–591.
- Morrell, C.N.; Aggrey, A.A.; Chapman, L.M.; Modjeski, K.L. Emerging Roles for Platelets as Immune and Inflammatory Cells. Blood 2014, 123, 2759–2767.
- Thachil, J. What Do Monitoring Platelet Counts in COVID-19 Teach Us? J. Thromb. Haemost. 2020, 18, 2071–2072.
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.-H.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.D.; Burns, A.J.; et al. Endotheliopathy in COVID-19-Associated Coagulopathy: Evidence from a Single-Centre, Cross-Sectional Study. Lancet Haematol. 2020, 7, e575–e582.
- Conti, P.; Caraffa, A.; Gallenga, C.E.; Ross, R.; Kritas, S.K.; Frydas, I.; Younes, A.; Di Emidio, P.; Ronconi, G.; Toniato, E. IL-1 Induces Throboxane-A2 (TxA2) in COVID-19 Causing Inflammation and Micro-Thrombi: Inhibitory Effect of the IL-1 Receptor Antagonist (IL-1Ra). J. Biol. Regul. Homeost. Agents 2020, 34, 1623–1627.
- Derhaschnig, U.; Schweeger-Exeli, I.; Marsik, C.; Cardona, F.; Minuz, P.; Jilma, B. Effects of Aspirin and NO-Aspirin (NCX 4016) on Platelet Function and Coagulation in Human Endotoxemia. Platelets 2010, 21, 320–328.
- Grobler, C.; Maphumulo, S.C.; Grobbelaar, L.M.; Bredenkamp, J.C.; Laubscher, G.J.; Lourens, P.J.; Steenkamp, J.; Kell, D.B.; Pretorius, E. COVID-19: The Rollercoaster of Fibrin(Ogen), D-Dimer, Von Willebrand Factor, P-Selectin and Their Interactions with Endothelial Cells, Platelets and Erythrocytes. Int. J. Mol. Sci. 2020, 21, 5168.

