COVID-19 infection is associated with a broad spectrum of presentations, but alveolar capillary microthrombi have been described as a common finding in COVID-19 patients, appearing as a consequence of a severe endothelial injury with endothelial cell membrane disruption. These observations clearly point to the identification of a COVID-19-associated coagulopathy, which may contribute to thrombosis, multi-organ damage, and cause of severity and fatality. One significant finding that emerges in prothrombotic abnormalities observed in COVID-19 patients is that the coagulation alterations are mainly mediated by the activation of platelets and intrinsically related to viral-mediated endothelial inflammation. Beyond the well-known role in hemostasis, the ability of platelets to also release various potent cytokines and chemokines has elevated these small cells from simple cell fragments to crucial modulators in the blood, including their inflammatory functions, that have a large influence on the immune response during infectious disease. Indeed, platelets are involved in the pathogenesis of acute lung injury also by promoting NET formation and affecting vascular permeability. Specifically, the deposition by activated platelets of the chemokine platelet factor 4 at sites of inflammation promotes adhesion of neutrophils on endothelial cells and thrombogenesis, and it seems deeply involved in the phenomenon of vaccine-induced thrombocytopenia and thrombosis. Importantly, the hyperactivated platelet phenotype along with evidence of cytokine storm, high levels of P-selectin, D-dimer, and, on the other hand, decreased levels of fibrinogen, von Willebrand factor, and thrombocytopenia may be considered suitable biomarkers that distinguish the late stage of COVID-19 progression in critically ill patients.
- COVID-19
- SARS-CoV-2
- thrombosis
1. Introduction
62. SARS-CoV-2 Effects on Platelets
Blood from patients with COVID-19 contains SARS-CoV-2, is infectious [104[18][19][20],105,106], and high mRNA levels of SARS-CoV-2 correlate with severity of infection [106,107][20][21]. In viral infections, such as influenza, human immunodeficiency, and hepatitis C, platelets internalize virions, resulting in platelet activation [52,108][22][23]. A direct action of SARS-CoV-2 on platelets is still controversial. In a study by Barrett et al., SARS-CoV-2 viral particles were found in megakaryocytes morphologically active in platelet production in the bone marrow [17] and within lung in deceased COVID-19 patients [17]. Consistently, viral particles were also found in platelets in approximately 39% of circulating platelets not showing changes in their morphology. Obviously, this suggests that SARS-CoV-2 is either transferred from megakaryocytes to platelets or directly engulfed by circulating platelets. The mechanism by which megakaryocytes and platelets take up SARS-CoV-2 has been explored and platelets have been reported to express the receptor for SARS-CoV-2 angiotensin-converting enzyme (ACE) 2 [105,106][19][20]. It is known that SARS-CoV-2 binds to and enters through cells that express ACE2 [109][24] and promotes an immediate downregulation of this receptor [110][25]. Virus binding to ACE2 receptor determines an increase of its substrate Angiotensin II (Ang II) with an impact on immune, vascular endothelial and coagulation responses [111][26]. Actually, as a result of Ang II accumulation secondary to ACE2 downregulation, clot formation may also occur for the consequent overexpression of TF [112][27], a transmembrane protein serving as a high affinity receptor and cofactor for coagulation factors VII and VIIa [113,114,115][28][29][30]. TF expression is negligible in healthy non-inflamed endothelial cells, but it can be induced by a number of proinflammatory stimuli including viruses [116,117,118][31][32][33]. TF binding to factor VII initiates the extrinsic coagulation cascade with generation of factor Xa and thrombin [115][30] which, in turn, activates platelets and triggers the conversion of fibrinogen to fibrin, essential for blood clotting [119][34]. COVID-19 patients show elevated TF activity in circulating extracellular vesicles associated with disease severity and mortality [120][35]. Pulmonary histopathology studies with the characterization of CD61+ platelet thrombi in COVID-19 patients with ARDS showed that CD61+ areas were higher in COVID-19 vs. non-COVID-19 ARDS samples [121][36]. Interestingly, the same authors found that higher levels of fibrin and activated platelets in PF4-positive thrombi correlated to high TF protein expression throughout lung tissue samples in which both arterial and venous thrombi and microangiopathy were observed [121][36]. Traces of SARS-CoV-2 mRNA, detected by reverse transcription quantitative real-time PCR (RT-qPCR), were found in isolated platelets in some studies [16] but not in all [122][37]. Furthermore, it has been found that SARS-CoV-2 mRNA can entry platelets also through mechanisms independent of ACE2 receptor [16]. In any case, platelet hyperactivation in COVID-19 patients has been documented by several studies [16,21,106,123,124][16][38][20][39][40] and increased aggregation, alpha-granule secretion, and thrombus formation seem to be also induced by a spike (S) protein fragment binding to platelets [105][19]. Indeed, SARS-CoV-2 S protein is formed by protruding homotrimers that play a key role in virus attachment to ACE2 receptor of target cells [125][41]. However, receptor binding per se could not explain each coagulopathy observed in patients affected by COVID-19. Actually, it has been found that S protein can be shed and free S protein subunits were detected in different organs and urine [126][42]. The coronavirus S glycoprotein is a class I viral fusion protein formed by S1 and S2 subunits [127][43]. The subunit S1 mediates receptor binding [128][44], while S2 is responsible for virus–cell membrane fusion [129][45]. In COVID-19 patients, free S1 particles have been detected in circulation and seem to be involved in the pathogenesis of the disease [130][46]. Interestingly, a confirmation of SARS-CoV-2 effects on platelets comes from a study carried out by Grobbelaar et al., who showed the in vitro ability of S protein to directly interact with platelets and fibrinogen to cause blood hypercoagulation [126][42]. Specifically, after that whole blood samples from healthy subjects were exposed to isolated SARS-CoV-2 S protein S1 subunit, platelet hyperactivation and major ultrastructural changes were noted. The first communication between hemostasis and inflammation occurs at the level of endothelium. Under physiological conditions, the balance between pro- and anti-thrombotic factors released by endothelial cells preserves an intact endothelium; conversely, if endothelium is damaged, it loses its anti-inflammatory and anti-thrombotic properties becoming suitable for inflammatory and prothrombotic environment [8,11,12,131][8][11][12][47]. While platelets physiologically contribute to guarantee the integrity of basal barrier of the alveolar capillaries, they may also play an important role in lung injury in a variety of pulmonary disorders [132][48]. The involvement of platelet–leukocyte aggregates and platelet–endothelial interactions in the pathogenesis of acute lung injury has already been observed [133][49]. As already mentioned, during SARS-CoV-2 infection, lung tissue injury and damage of lung endothelial cells promote platelet aggregation with the formation of microthrombi and consequent consumption of platelets [134][50]. A number of circulating and dysregulated coagulation and inflammation biomarkers, including D-dimer, P-selectin, fibrinogen, and VWF and various cytokines, can directly bind to endothelial cell receptors thus influencing signaling pathways involved in endotheliopathy [1,135][1][51]. Certainly, it has to be considered that platelet hyperactivation may be also the consequence of a damaged endothelium as well as of the cytokine storm occurring during SARS-CoV-2 infection [136][52]. Actually, alterations in endothelial cell functions cause the decreased production of molecules, such as nitric oxide and prostacyclin, known to prevent platelet adhesion and the increased secretion of platelet activators resulting in platelet hyperactivation [137][53]. Clinical observations may induce to hypothesize that in COVID-19 patients the measurement of D-dimer, fibrinogen, VWF, and the platelet activation marker P-selectin may help clinicians in deciding treatment strategy on the basis of correct clinical diagnosis [138][54]. Specifically, as shown in Figure 31, during early stages, COVID-19 patients show normal to slightly enhanced levels of D-dimer, fibrinogen, VWF and P-selectin, and platelet activation. If untreated, D-dimer rapidly increases and also fibrinogen, VWF and P-selectin further increase leading to platelet hyperactivation, clot formation, and thrombotic events. During the late stage of the disease, critically ill patients show cytokine storm, still high levels of P-selectin and D-dimer, while fibrinogen and VWF decrease because they are depleted by damaged endothelial cells or hyperactivated platelets that, at this stage, show thrombocytopenia (Figure 31).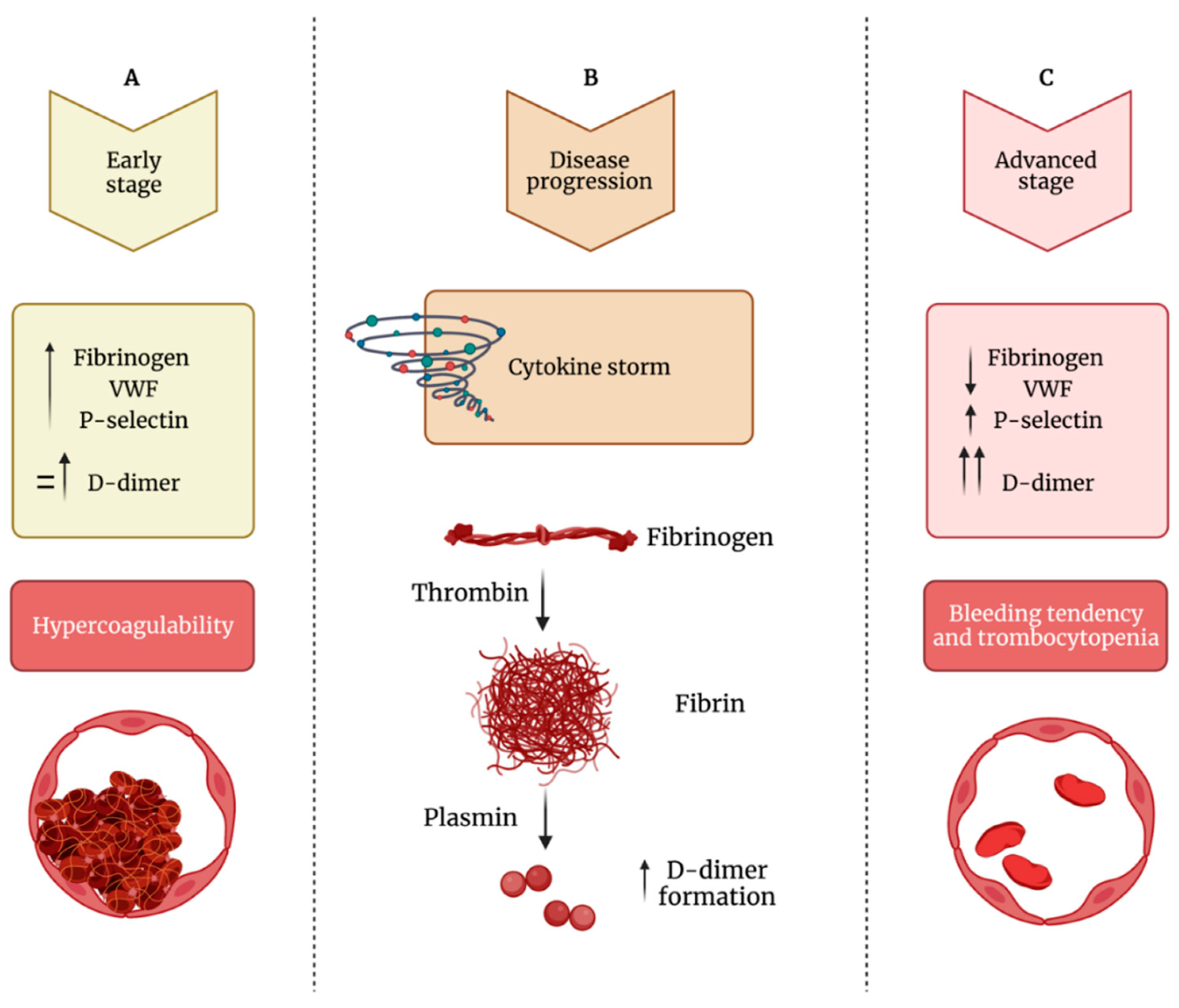 Figure 31. Impaired balance between hypercoagulability and bleeding tendency during COVID-19 progression. (A) During the early phase of the infection, patients show increased levels of fibrinogen, VWF (von Willebrand factor) and P-selectin and normal to mildly increased levels of D-dimer, leading to hypercoagulability. (B) During the disease’s progression, there is increased formation of D-dimer. (C) In the advanced stage of the infection, fibrinogen and VWF levels decrease, while D-dimer levels strongly increased; this phase is characterized by thrombocytopenia and bleeding diathesis.
Figure 31. Impaired balance between hypercoagulability and bleeding tendency during COVID-19 progression. (A) During the early phase of the infection, patients show increased levels of fibrinogen, VWF (von Willebrand factor) and P-selectin and normal to mildly increased levels of D-dimer, leading to hypercoagulability. (B) During the disease’s progression, there is increased formation of D-dimer. (C) In the advanced stage of the infection, fibrinogen and VWF levels decrease, while D-dimer levels strongly increased; this phase is characterized by thrombocytopenia and bleeding diathesis.
3. Conclusions
References
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128.
- Yeaman, M.R. Bacterial-Platelet Interactions: Virulence Meets Host Defense. Future Microbiol. 2010, 5, 471–506.
- De Stoppelaar, S.F.; van’t Veer, C.; van der Poll, T. The Role of Platelets in Sepsis. Thromb. Haemost. 2014, 112, 666–677.
- Jenne, C.N.; Kubes, P. Platelets in Inflammation and Infection. Platelets 2015, 26, 286–292.
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basílio, J.; Petzelbauer, P.; Assinger, A.; et al. Cell Type-Specific Roles of NF-ΚB Linking Inflammation and Thrombosis. Front. Immunol. 2019, 10, 85.
- Liverani, E.; Mondrinos, M.J.; Sun, S.; Kunapuli, S.P.; Kilpatrick, L.E. Role of Protein Kinase C-Delta in Regulating Platelet Activation and Platelet-Leukocyte Interaction during Sepsis. PLoS ONE 2018, 13, e0195379.
- Acanfora, D.; Acanfora, C.; Ciccone, M.M.; Scicchitano, P.; Bortone, A.S.; Uguccioni, M.; Casucci, G. The Cross-Talk between Thrombosis and Inflammatory Storm in Acute and Long-COVID-19: Therapeutic Targets and Clinical Cases. Viruses 2021, 13, 1904.
- Cicala, C.; Cirino, G. Linkage between Inflammation and Coagulation: An Update on the Molecular Basis of the Crosstalk. Life Sci. 1998, 62, 1817–1824.
- Esmon, C.T.; Xu, J.; Lupu, F. Innate Immunity and Coagulation. J. Thromb. Haemost. 2011, 9 (Suppl. 1), 182–188.
- Taus, F.; Salvagno, G.; Canè, S.; Fava, C.; Mazzaferri, F.; Carrara, E.; Petrova, V.; Barouni, R.M.; Dima, F.; Dalbeni, A.; et al. Platelets Promote Thromboinflammation in SARS-CoV-2 Pneumonia. Arter. Thromb. Vasc. Biol. 2020, 40, 2975–2989.
- Hansson, G.K.; Robertson, A.-K.L.; Söderberg-Nauclér, C. Inflammation and Atherosclerosis. Annu. Rev. Pathol. 2006, 1, 297–329.
- Stark, K.; Massberg, S. Interplay between Inflammation and Thrombosis in Cardiovascular Pathology. Nat. Rev. Cardiol. 2021, 18, 666–682.
- McFadyen, J.D.; Stevens, H.; Peter, K. The Emerging Threat of (Micro)Thrombosis in COVID-19 and Its Therapeutic Implications. Circ. Res. 2020, 127, 571–587.
- Melillo, F.; Napolano, A.; Loffi, M.; Regazzoni, V.; Boccellino, A.; Danzi, G.B.; Cappelletti, A.M.; Rovere-Querini, P.; Landoni, G.; Ingallina, G.; et al. Myocardial Injury in Patients with SARS-CoV-2 Pneumonia: Pivotal Role of Inflammation in COVID-19. Eur. J. Clin. Investig. 2021, e13703.
- Attiq, A.; Yao, L.J.; Afzal, S.; Khan, M.A. The Triumvirate of NF-ΚB, Inflammation and Cytokine Storm in COVID-19. Int. Immunopharmacol. 2021, 101, 108255.
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet Gene Expression and Function in Patients with COVID-19. Blood 2020, 136, 1317–1329.
- Barrett, T.J.; Bilaloglu, S.; Cornwell, M.; Burgess, H.M.; Virginio, V.W.; Drenkova, K.; Ibrahim, H.; Yuriditsky, E.; Aphinyanaphongs, Y.; Lifshitz, M.; et al. Platelets Contribute to Disease Severity in COVID-19. J. Thromb. Haemost. 2021.
- Wang, W.; Xu, Y.; Gao, R.; Lu, R.; Han, K.; Wu, G.; Tan, W. Detection of SARS-CoV-2 in Different Types of Clinical Specimens. JAMA 2020, 323, 1843–1844.
- Campbell, R.A.; Boilard, E.; Rondina, M.T. Is There a Role for the ACE2 Receptor in SARS-CoV-2 Interactions with Platelets? J. Thromb. Haemost. 2021, 19, 46–50.
- Zhang, S.; Liu, Y.; Wang, X.; Yang, L.; Li, H.; Wang, Y.; Liu, M.; Zhao, X.; Xie, Y.; Yang, Y.; et al. SARS-CoV-2 Binds Platelet ACE2 to Enhance Thrombosis in COVID-19. J. Hematol. Oncol. 2020, 13, 120.
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical Features of Patients Infected with 2019 Novel Coronavirus in Wuhan, China. Lancet 2020, 395, 497–506.
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circ. Res. 2018, 122, 337–351.
- Koupenova, M.; Freedman, J.E. Platelets: The Unsung Hero of the Immune Response. J. Thromb. Haemost. 2015, 13, 268–270.
- Ding, Y.; Wang, H.; Shen, H.; Li, Z.; Geng, J.; Han, H.; Cai, J.; Li, X.; Kang, W.; Weng, D.; et al. The Clinical Pathology of Severe Acute Respiratory Syndrome (SARS): A Report from China. J. Pathol. 2003, 200, 282–289.
- Malha, L.; Mueller, F.B.; Pecker, M.S.; Mann, S.J.; August, P.; Feig, P.U. COVID-19 and the Renin-Angiotensin System. Kidney Int. Rep. 2020, 5, 563–565.
- Harrison, D.G.; Guzik, T.J.; Lob, H.E.; Madhur, M.S.; Marvar, P.J.; Thabet, S.R.; Vinh, A.; Weyand, C.M. Inflammation, Immunity, and Hypertension. Hypertension 2011, 57, 132–140.
- Dmitrieva, N.I.; Burg, M.B. Elevated Sodium and Dehydration Stimulate Inflammatory Signaling in Endothelial Cells and Promote Atherosclerosis. PLoS ONE 2015, 10, e0128870.
- Nemerson, Y. Tissue Factor and Hemostasis. Blood 1988, 71, 1–8.
- Mackman, N.; Tilley, R.E.; Key, N.S. Role of the Extrinsic Pathway of Blood Coagulation in Hemostasis and Thrombosis. Arter. Thromb. Vasc. Biol. 2007, 27, 1687–1693.
- Grover, S.P.; Mackman, N. Tissue Factor: An Essential Mediator of Hemostasis and Trigger of Thrombosis. Arter. Thromb. Vasc. Biol. 2018, 38, 709–725.
- Mackman, N. Regulation of the Tissue Factor Gene. Thromb. Haemost. 1997, 78, 747–754.
- Wang, J.; Mahmud, S.A.; Bitterman, P.B.; Huo, Y.; Slungaard, A. Histone Deacetylase Inhibitors Suppress TF-KappaB-Dependent Agonist-Driven Tissue Factor Expression in Endothelial Cells and Monocytes. J. Biol. Chem. 2007, 282, 28408–28418.
- Li, Y.-D.; Ye, B.-Q.; Zheng, S.-X.; Wang, J.-T.; Wang, J.-G.; Chen, M.; Liu, J.-G.; Pei, X.-H.; Wang, L.-J.; Lin, Z.-X.; et al. NF-KappaB Transcription Factor P50 Critically Regulates Tissue Factor in Deep Vein Thrombosis. J. Biol. Chem. 2009, 284, 4473–4483.
- Mackman, N. The Many Faces of Tissue Factor. J. Thromb. Haemost. 2009, 7 (Suppl. 1), 136–139.
- Rosell, A.; Havervall, S.; von Meijenfeldt, F.; Hisada, Y.; Aguilera, K.; Grover, S.P.; Lisman, T.; Mackman, N.; Thålin, C. Patients With COVID-19 Have Elevated Levels of Circulating Extracellular Vesicle Tissue Factor Activity That Is Associated with Severity and Mortality-Brief Report. Arter. Thromb. Vasc. Biol. 2021, 41, 878–882.
- Subrahmanian, S.; Borczuk, A.; Salvatore, S.; Fung, K.-M.; Merrill, J.T.; Laurence, J.; Ahamed, J. Tissue Factor Upregulation Is Associated with SARS-CoV-2 in the Lungs of COVID-19 Patients. J. Thromb. Haemost. 2021, 19, 2268–2274.
- Bury, L.; Camilloni, B.; Castronari, R.; Piselli, E.; Malvestiti, M.; Borghi, M.; KuchiBotla, H.; Falcinelli, E.; Petito, E.; Amato, F.; et al. Search for SARS-CoV-2 RNA in Platelets from COVID-19 Patients. Platelets 2021, 32, 284–287.
- Zaid, Y.; Puhm, F.; Allaeys, I.; Naya, A.; Oudghiri, M.; Khalki, L.; Limami, Y.; Zaid, N.; Sadki, K.; Ben, E.l.; et al. Platelets Can Associate with SARS-Cov-2 RNA and Are Hyperactivated in COVID-19. Circ. Res. 2020, 127, 1404–1418.
- Barrett, T.J.; Lee, A.H.; Xia, Y.; Lin, L.H.; Black, M.; Cotzia, P.; Hochman, J.; Berger, J.S. Platelet and Vascular Biomarkers Associate with Thrombosis and Death in Coronavirus Disease. Circ. Res. 2020, 127, 945–947.
- Shen, B.; Yi, X.; Sun, Y.; Bi, X.; Du, J.; Zhang, C.; Quan, S.; Zhang, F.; Sun, R.; Qian, L.; et al. Proteomic and Metabolomic Characterization of COVID-19 Patient Sera. Cell 2020, 182, 59–72.e15.
- Bergmann, C.C.; Silverman, R.H. COVID-19: Coronavirus Replication, Pathogenesis, and Therapeutic Strategies. Clevel. Clin. J. Med. 2020, 87, 321–327.
- Grobbelaar, L.M.; Venter, C.; Vlok, M.; Ngoepe, M.; Laubscher, G.J.; Lourens, P.J.; Steenkamp, J.; Kell, D.B.; Pretorius, E. SARS-CoV-2 Spike Protein S1 Induces Fibrin(Ogen) Resistant to Fibrinolysis: Implications for Microclot Formation in COVID-19. Biosci. Rep. 2021, 41, BSR20210611.
- Kawase, M.; Kataoka, M.; Shirato, K.; Matsuyama, S. Biochemical Analysis of Coronavirus Spike Glycoprotein Conformational Intermediates during Membrane Fusion. J. Virol. 2019, 93, e00785-19.
- Watanabe, Y.; Allen, J.D.; Wrapp, D.; McLellan, J.S.; Crispin, M. Site-Specific Glycan Analysis of the SARS-CoV-2 Spike. Science 2020, 369, 330–333.
- Flores-Alanis, A.; Sandner-Miranda, L.; Delgado, G.; Cravioto, A.; Morales-Espinosa, R. The Receptor Binding Domain of SARS-CoV-2 Spike Protein Is the Result of an Ancestral Recombination between the Bat-CoV RaTG13 and the Pangolin-CoV MP789. BMC Res. Notes 2020, 13, 398.
- Letarov, A.V.; Babenko, V.V.; Kulikov, E.E. Free SARS-CoV-2 Spike Protein S1 Particles May Play a Role in the Pathogenesis of COVID-19 Infection. Biochemistry 2021, 86, 257–261.
- Engelmann, B.; Massberg, S. Thrombosis as an Intravascular Effector of Innate Immunity. Nat. Rev. Immunol. 2013, 13, 34–45.
- Weyrich, A.S.; Zimmerman, G.A. Platelets in Lung Biology. Annu. Rev. Physiol. 2013, 75, 569–591.
- Morrell, C.N.; Aggrey, A.A.; Chapman, L.M.; Modjeski, K.L. Emerging Roles for Platelets as Immune and Inflammatory Cells. Blood 2014, 123, 2759–2767.
- Thachil, J. What Do Monitoring Platelet Counts in COVID-19 Teach Us? J. Thromb. Haemost. 2020, 18, 2071–2072.
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.-H.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.D.; Burns, A.J.; et al. Endotheliopathy in COVID-19-Associated Coagulopathy: Evidence from a Single-Centre, Cross-Sectional Study. Lancet Haematol. 2020, 7, e575–e582.
- Conti, P.; Caraffa, A.; Gallenga, C.E.; Ross, R.; Kritas, S.K.; Frydas, I.; Younes, A.; Di Emidio, P.; Ronconi, G.; Toniato, E. IL-1 Induces Throboxane-A2 (TxA2) in COVID-19 Causing Inflammation and Micro-Thrombi: Inhibitory Effect of the IL-1 Receptor Antagonist (IL-1Ra). J. Biol. Regul. Homeost. Agents 2020, 34, 1623–1627.
- Derhaschnig, U.; Schweeger-Exeli, I.; Marsik, C.; Cardona, F.; Minuz, P.; Jilma, B. Effects of Aspirin and NO-Aspirin (NCX 4016) on Platelet Function and Coagulation in Human Endotoxemia. Platelets 2010, 21, 320–328.
- Grobler, C.; Maphumulo, S.C.; Grobbelaar, L.M.; Bredenkamp, J.C.; Laubscher, G.J.; Lourens, P.J.; Steenkamp, J.; Kell, D.B.; Pretorius, E. COVID-19: The Rollercoaster of Fibrin(Ogen), D-Dimer, Von Willebrand Factor, P-Selectin and Their Interactions with Endothelial Cells, Platelets and Erythrocytes. Int. J. Mol. Sci. 2020, 21, 5168.