Given the knowledge we have, there is still no clarity on the pathophysiological mechanisms involved in arterial and venous thrombosis during COVID-19 disease and, particularly, some aspects of platelet susceptibility to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) are in contrast. However, the literature agrees on the occurrence of prothrombotic abnormalities and, in this scenario, platelets have been shown to be deeply involved. Traditionally platelets are recognized as key actors in hemostasis. Indeed, we have to consider that platelets are also deeply implicated in the host defense in case of infections [
2] given that, together with other immune cells and coagulation process, they act as modulators and effectors of immune cells other than clot formation [
3]. In vertebrates, platelets are able to respond to pathogens mainly by promoting neutrophil extracellular traps (NETs) release and indirectly disposing of them [
4]. The ability to prime macrophages, recruit and activate neutrophils, and participate actively to intravascular thrombosis places platelets as a link between host defense and thromboinflammation. Indeed, inflammation may cause hemostatic alterations leading to thrombosis [
5,
6] and, on the other hand, thrombosis may exacerbate inflammation [
7], thus promoting a loop which increases tissue damage and thrombotic complications [
8,
9].
6. SARS-CoV-2 Effects on Platelets
Blood from patients with COVID-19 contains SARS-CoV-2, is infectious [
104,
105,
106], and high mRNA levels of SARS-CoV-2 correlate with severity of infection [
106,
107].
In viral infections, such as influenza, human immunodeficiency, and hepatitis C, platelets internalize virions, resulting in platelet activation [
52,
108]. A direct action of SARS-CoV-2 on platelets is still controversial. In a study by Barrett et al., SARS-CoV-2 viral particles were found in megakaryocytes morphologically active in platelet production in the bone marrow [
17] and within lung in deceased COVID-19 patients [
17]. Consistently, viral particles were also found in platelets in approximately 39% of circulating platelets not showing changes in their morphology. Obviously, this suggests that SARS-CoV-2 is either transferred from megakaryocytes to platelets or directly engulfed by circulating platelets.
The mechanism by which megakaryocytes and platelets take up SARS-CoV-2 has been explored and platelets have been reported to express the receptor for SARS-CoV-2 angiotensin-converting enzyme (ACE) 2 [
105,
106]. It is known that SARS-CoV-2 binds to and enters through cells that express ACE2 [
109] and promotes an immediate downregulation of this receptor [
110]. Virus binding to ACE2 receptor determines an increase of its substrate Angiotensin II (Ang II) with an impact on immune, vascular endothelial and coagulation responses [
111]. Actually, as a result of Ang II accumulation secondary to ACE2 downregulation, clot formation may also occur for the consequent overexpression of TF [
112], a transmembrane protein serving as a high affinity receptor and cofactor for coagulation factors VII and VIIa [
113,
114,
115].
TF expression is negligible in healthy non-inflamed endothelial cells, but it can be induced by a number of proinflammatory stimuli including viruses [
116,
117,
118].
TF binding to factor VII initiates the extrinsic coagulation cascade with generation of factor Xa and thrombin [
115] which, in turn, activates platelets and triggers the conversion of fibrinogen to fibrin, essential for blood clotting [
119]. COVID-19 patients show elevated TF activity in circulating extracellular vesicles associated with disease severity and mortality [
120]. Pulmonary histopathology studies with the characterization of CD61+ platelet thrombi in COVID-19 patients with ARDS showed that CD61+ areas were higher in COVID-19 vs. non-COVID-19 ARDS samples [
121]. Interestingly, the same authors found that higher levels of fibrin and activated platelets in PF4-positive thrombi correlated to high TF protein expression throughout lung tissue samples in which both arterial and venous thrombi and microangiopathy were observed [
121].
Traces of SARS-CoV-2 mRNA, detected by reverse transcription quantitative real-time PCR (RT-qPCR), were found in isolated platelets in some studies [
16] but not in all [
122]. Furthermore, it has been found that SARS-CoV-2 mRNA can entry platelets also through mechanisms independent of ACE2 receptor [
16].
In any case, platelet hyperactivation in COVID-19 patients has been documented by several studies [
16,
21,
106,
123,
124] and increased aggregation, alpha-granule secretion, and thrombus formation seem to be also induced by a spike (S) protein fragment binding to platelets [
105]. Indeed, SARS-CoV-2 S protein is formed by protruding homotrimers that play a key role in virus attachment to ACE2 receptor of target cells [
125]. However, receptor binding per se could not explain each coagulopathy observed in patients affected by COVID-19. Actually, it has been found that S protein can be shed and free S protein subunits were detected in different organs and urine [
126]. The coronavirus S glycoprotein is a class I viral fusion protein formed by S1 and S2 subunits [
127]. The subunit S1 mediates receptor binding [
128], while S2 is responsible for virus–cell membrane fusion [
129]. In COVID-19 patients, free S1 particles have been detected in circulation and seem to be involved in the pathogenesis of the disease [
130]. Interestingly, a confirmation of SARS-CoV-2 effects on platelets comes from a study carried out by Grobbelaar et al., who showed the in vitro ability of S protein to directly interact with platelets and fibrinogen to cause blood hypercoagulation [
126]. Specifically, after that whole blood samples from healthy subjects were exposed to isolated SARS-CoV-2 S protein S1 subunit, platelet hyperactivation and major ultrastructural changes were noted.
The first communication between hemostasis and inflammation occurs at the level of endothelium. Under physiological conditions, the balance between pro- and anti-thrombotic factors released by endothelial cells preserves an intact endothelium; conversely, if endothelium is damaged, it loses its anti-inflammatory and anti-thrombotic properties becoming suitable for inflammatory and prothrombotic environment [
8,
11,
12,
131]. While platelets physiologically contribute to guarantee the integrity of basal barrier of the alveolar capillaries, they may also play an important role in lung injury in a variety of pulmonary disorders [
132]. The involvement of platelet–leukocyte aggregates and platelet–endothelial interactions in the pathogenesis of acute lung injury has already been observed [
133].
As already mentioned, during SARS-CoV-2 infection, lung tissue injury and damage of lung endothelial cells promote platelet aggregation with the formation of microthrombi and consequent consumption of platelets [
134]. A number of circulating and dysregulated coagulation and inflammation biomarkers, including D-dimer, P-selectin, fibrinogen, and VWF and various cytokines, can directly bind to endothelial cell receptors thus influencing signaling pathways involved in endotheliopathy [
1,
135]. Certainly, it has to be considered that platelet hyperactivation may be also the consequence of a damaged endothelium as well as of the cytokine storm occurring during SARS-CoV-2 infection [
136]. Actually, alterations in endothelial cell functions cause the decreased production of molecules, such as nitric oxide and prostacyclin, known to prevent platelet adhesion and the increased secretion of platelet activators resulting in platelet hyperactivation [
137].
Clinical observations may induce to hypothesize that in COVID-19 patients the measurement of D-dimer, fibrinogen, VWF, and the platelet activation marker P-selectin may help clinicians in deciding treatment strategy on the basis of correct clinical diagnosis [
138]. Specifically, as shown in
Figure 3, during early stages, COVID-19 patients show normal to slightly enhanced levels of D-dimer, fibrinogen, VWF and P-selectin, and platelet activation. If untreated, D-dimer rapidly increases and also fibrinogen, VWF and P-selectin further increase leading to platelet hyperactivation, clot formation, and thrombotic events. During the late stage of the disease, critically ill patients show cytokine storm, still high levels of P-selectin and D-dimer, while fibrinogen and VWF decrease because they are depleted by damaged endothelial cells or hyperactivated platelets that, at this stage, show thrombocytopenia (
Figure 3).
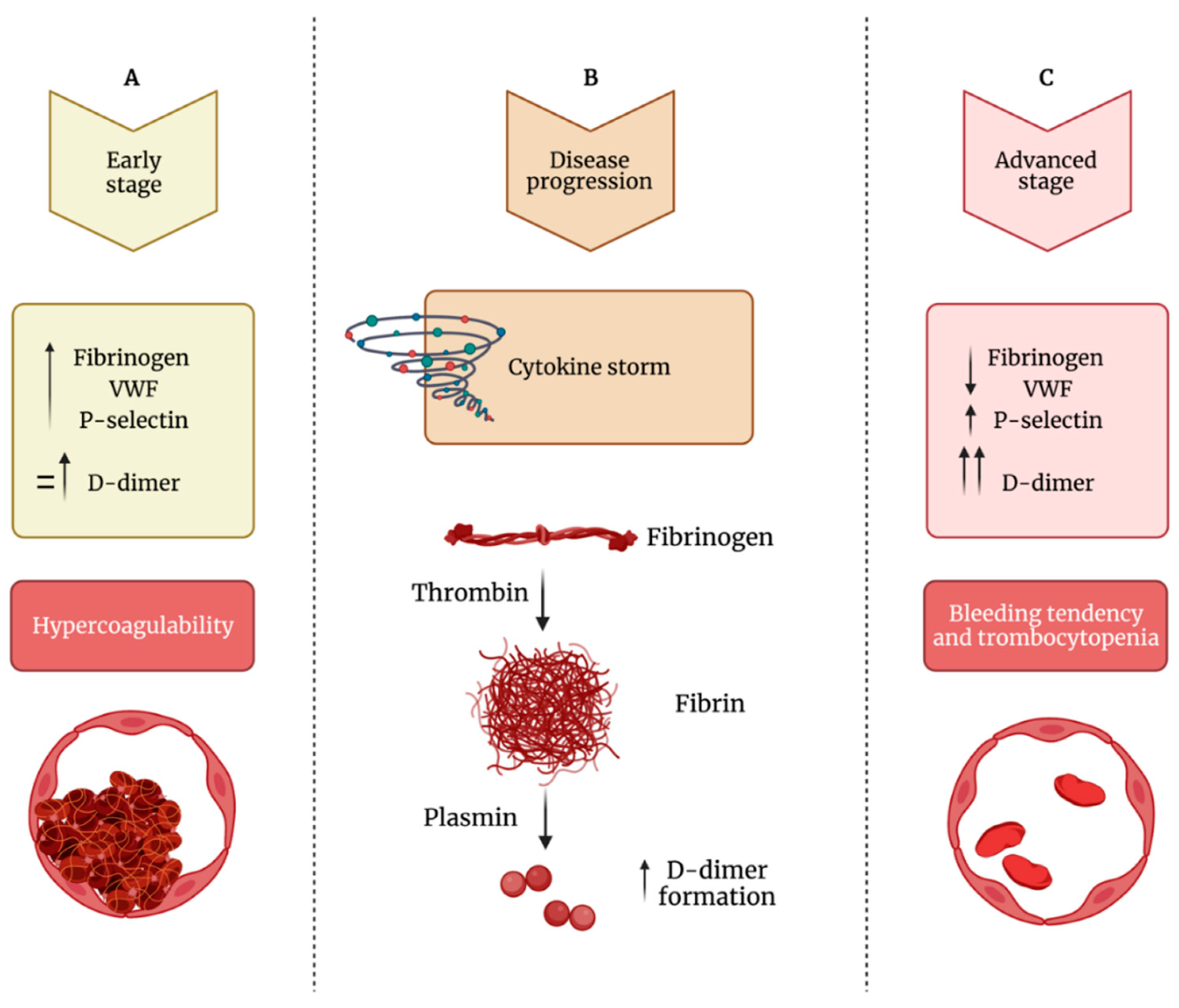
Figure 3. Impaired balance between hypercoagulability and bleeding tendency during COVID-19 progression. (A) During the early phase of the infection, patients show increased levels of fibrinogen, VWF (von Willebrand factor) and P-selectin and normal to mildly increased levels of D-dimer, leading to hypercoagulability. (B) During the disease’s progression, there is increased formation of D-dimer. (C) In the advanced stage of the infection, fibrinogen and VWF levels decrease, while D-dimer levels strongly increased; this phase is characterized by thrombocytopenia and bleeding diathesis.
3. Conclusions
Several independent studies are in line to consider platelets the frontline of COVID-19 pathogenesis for their involvement in different stages of SARS-CoV-2 infection. The most common complication in patients severely affected by COVID-19 is thrombocytopenia, basically related to decreased platelet production and increased platelet consumption and disruption.
However, platelet abnormalities in COVID-19 are not only quantitative. Indeed, it has been clearly established that platelets are also hyperreactive, thus consistently contributing to the overwhelming of thromboinflammatory events. Moreover, it cannot be excluded that platelets may also work as a reservoir for SARS-CoV-2 infection, replication, and spread. Indeed, further studies are needed to clarify these potential aspects of platelets versus the virion responsible for COVID-19. Additionally, not only ACE2 but also other platelet receptors have been reported to regulate SARS-CoV-2 engagement of platelets. In any case, platelets participate actively to intravascular thrombosis representing a link between host defense and thromboinflammation, a process in which their interactions with activated neutrophils, monocytes, coagulation systems and endothelial cells lead to intravascular clot formation from small to large vessels. Certainly, platelet potential to contribute to the overwhelming thromboinflammatory responses might suggest that a tailored antiplatelet therapy in addition to heparin could improve the outcomes during COVID-19. Further exploration of this pharmacological strategy needs to be done.