 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Matthew Asmussen | + 1980 word(s) | 1980 | 2021-12-15 04:57:01 | | | |
| 2 | Yvaine Wei | -11 word(s) | 1969 | 2021-12-28 02:40:22 | | |
Video Upload Options
Electrocatalytic water splitting is a possible route to the expanded generation of green hydrogen; a long-term challenge is the requirement of fresh water as an electrolyzer feed. The use of seawater as a direct feed for electrolytic hydrogen production would alleviate fresh water needs and potentially open an avenue for locally generated hydrogen from marine hydrokinetic or off-shore power sources. One environmental limitation to seawater electrolysis is the generation of chlorine as a competitive anodic reaction.
1. Introduction
2. Electrode Fabrication and Structure
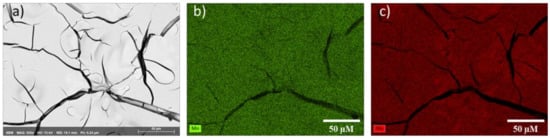

| Spectrum | O (at%) | Mn (at%) | Mo (at%) | Ru (at%) |
|---|---|---|---|---|
| 1 | 58.23 | 41.49 | 0.28 | 0.00 |
| 2 | 58.17 | 41.29 | 0.40 | 0.15 |
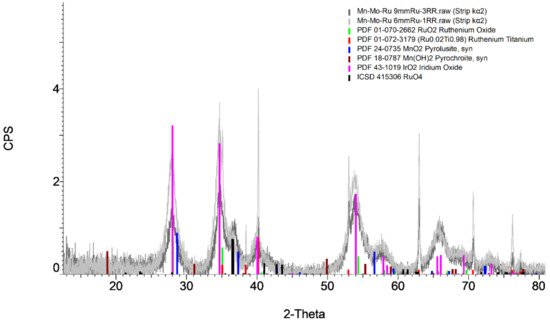
3. OER Efficiency in Saline and Seawater Environments
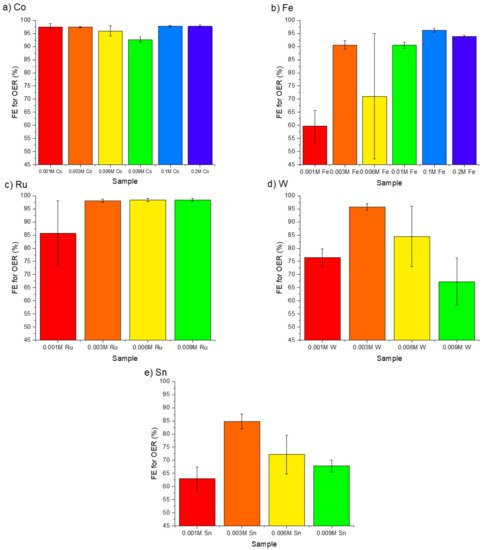
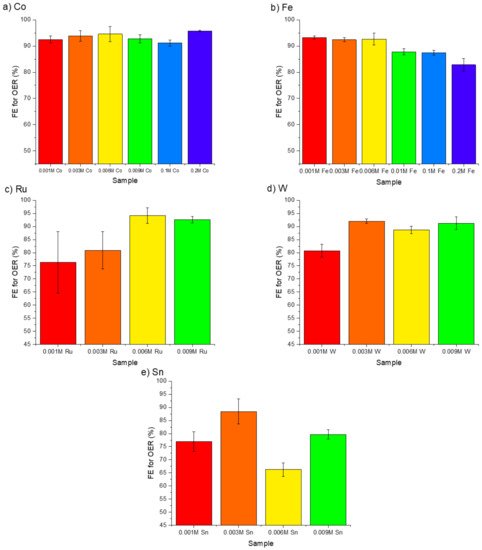
4. The Effectiveness of Doping Transition Metals on CER Suppression
The doping transition metals into Mn-Mo-X catalysts can effectively suppress the CER and improve overall catalytic activity. The best-performing dopant was Ru in terms of efficiency for OER over CER and catalytic activity. However, despite previous reports of this doping approach being successful for CER suppression, little mechanistic discussion is present in the literature. With the similar response of multiple dopants (Fe, Sn, W, and Co) and the enhanced activity of a known catalytically active species, Ru, dopants may play a role in manipulating the coating density and stability in addition to serving as active catalytic sites. Evidence on both mechanistic roles exists. Ru and Ir are industrial benchmark catalysts for efficient oxygen and chlorine evolution (in the chlor-alkali process), sitting high on the volcano curve for both OER [27] and CER [28]. In the case of IrOx, the OER is not suppressed or influenced by the CER occurring on the same surface, showing scaling between the two reactions on active catalysts [29]. Targeting selective OER over CER can present a challenge but may be realized through a combination of processes.
The adsorbed intermediates generated for the CER can also influence selectivity. Surfaces that form a M-Cl intermediate can efficiently sustain the CER, while the generation of the M-OCl intermediate will suppress the CER [30]. The OCl intermediate route further suppresses the CER on RuO2 with increasing pH due to the change in Gibbs free energy loss. Studies of preferential OER in halide-containing environments on NiB and CoP catalysts suggest that the edge sites on the metalate clusters are crucial to sustaining the OER, as H2O will out compete halides for these sites, increasing the likelihood of OER over CER. With the small locations and disordered distribution of the Ru dopants, a higher number of edge sites may dictate an added preference to OER over CER via oxygen-containing intermediates [31].
References
- Tong, W.; Forster, M.; Dionigi, F.; Dresp, S.; Sadeghi Erami, R.; Strasser, P.; Cowan, A.J.; Farràs, P. Electrolysis of low-grade and saline surface water. Nat. Energy 2020, 5, 367–377.
- Webber, M.E. The water intensity of the transitional hydrogen economy. Environ. Res. Lett. 2007, 2, 034007.
- Dresp, S.R.; Dionigi, F.; Klingenhof, M.; Strasser, P. Direct electrolytic splitting of seawater: Opportunities and challenges. ACS Energy Lett. 2019, 4, 933–942.
- Hansen, H.A.; Man, I.A.; Studt, F.; Abild-Pedersen, F.; Bligaard, T.; Rossmeisl, J. Electrochemical chlorine evolution at rutile oxide (110) surfaces. Phys. Chem. Chem. Phys. 2010, 12, 283–290.
- El-Moneim, A.A.; Bhattarai, J.; Kato, Z.; Izumiya, K.; Kumagai, N.; Hashimoto, K. Mn-Mo-Sn oxide anodes for oxygen evolution in seawater electrolysis for hydrogen production. ECS Trans. 2010, 25, 127.
- El-Moneim, A.A.; Kumagai, N.; Asami, K.; Hashimoto, K. Nanocrystalline manganese-molybdenum-tungsten oxide anodes for oxygen evolution in acidic seawater electrolysis. Mater. Trans. 2005, 46, 309–316.
- Vos, J.; Koper, M. Measurement of competition between oxygen evolution and chlorine evolution using rotating ring-disk electrode voltammetry. J. Electroanal. Chem. 2018, 819, 260–268.
- Vos, J.G.; Wezendonk, T.A.; Jeremiasse, A.W.; Koper, M.T. MnOx/IrOx as selective oxygen evolution electrocatalyst in acidic chloride solution. J. Am. Chem. Soc. 2018, 140, 10270–10281.
- Amikam, G.; Nativ, P.; Gendel, Y. Chlorine-free alkaline seawater electrolysis for hydrogen production. Int. J. Hydrogen Energy 2018, 43, 6504–6514.
- Kuang, Y.; Kenney, M.J.; Meng, Y.; Hung, W.-H.; Liu, Y.; Huang, J.E.; Prasanna, R.; Li, P.; Li, Y.; Wang, L. Solar-driven, highly sustained splitting of seawater into hydrogen and oxygen fuels. Proc. Natl. Acad. Sci. USA 2019, 116, 6624–6629.
- Juodkazytė, J.; Šebeka, B.; Savickaja, I.; Petrulevičienė, M.; Butkutė, S.; Jasulaitienė, V.; Selskis, A.; Ramanauskas, R. Electrolytic splitting of saline water: Durable nickel oxide anode for selective oxygen evolution. Int. J. Hydrogen Energy 2019, 44, 5929–5939.
- Dresp, S.R.; Dionigi, F.; Klingenhof, M.; Merzdorf, T.; Schmies, H.; Drnec, J.; Poulain, A.; Strasser, P. Molecular Understanding of the Impact of Saline Contaminants and Alkaline pH on NiFe Layered Double Hydroxide Oxygen Evolution Catalysts. ACS Catal. 2021, 11, 6800–6809.
- Karlsson, R.K.; Cornell, A. Selectivity between oxygen and chlorine evolution in the chlor-alkali and chlorate processes. Chem. Rev. 2016, 116, 2982–3028.
- Exner, K.S.; Over, H. Beyond the rate-determining step in the oxygen evolution reaction over a single-crystalline IrO2 (110) model electrode: Kinetic scaling relations. ACS Catal. 2019, 9, 6755–6765.
- Exner, K.S. Controlling stability and selectivity in the competing chlorine and oxygen evolution reaction over transition metal oxide electrodes. ChemElectroChem 2019, 6, 3401–3409.
- Zheng, J. Seawater splitting for high-efficiency hydrogen evolution by alloyed PtNix electrocatalysts. Appl. Surf. Sci. 2017, 413, 360–365.
- Zheng, J. Pt-free NiCo electrocatalysts for oxygen evolution by seawater splitting. Electrochim. Acta 2017, 247, 381–391.
- Cheng, F.; Feng, X.; Chen, X.; Lin, W.; Rong, J.; Yang, W. Synergistic action of Co-Fe layered double hydroxide electrocatalyst and multiple ions of sea salt for efficient seawater oxidation at near-neutral pH. Electrochim. Acta 2017, 251, 336–343.
- Hsu, G.-S.W.; Lu, Y.-F.; Hsu, S.-Y. Effects of electrolysis time and electric potential on chlorine generation of electrolyzed deep ocean water. J. Food Drug Anal. 2017, 25, 759–765.
- Fujimura, K.; Izumiya, K.; Kawashima, A.; Akiyama, E.; Habazaki, H.; Kumagai, N.; Hashimoto, K. Anodically deposited manganese-molybdenum oxide anodes with high selectivity for evolving oxygen in electrolysis of seawater. J. Appl. Electrochem. 1999, 29, 769–775.
- Fujimura, K.; Matsui, T.; Izumiya, K.; Kumagai, N.; Akiyama, E.; Habazaki, H.; Kawashima, A.; Asami, K.; Hashimoto, K. Oxygen evolution on manganese–molybdenum oxide anodes in seawater electrolysis. Mater. Sci. Eng. A 1999, 267, 254–259.
- El-Moneim, A. Mn–Mo–W-oxide anodes for oxygen evolution during seawater electrolysis for hydrogen production: Effect of repeated anodic deposition. Int. J. Hydrogen Energy 2011, 36, 13398–13406.
- Dionigi, F.; Reier, T.; Pawolek, Z.; Gliech, M.; Strasser, P. Design criteria, operating conditions, and nickel-iron hydroxide catalyst materials for selective seawater electrolysis. ChemSusChem 2016, 9, 962–972.
- Vesborg, P.C.; Jaramillo, T.F. Addressing the terawatt challenge: Scalability in the supply of chemical elements for renewable energy. Rsc Adv. 2012, 2, 7933–7947.
- Kato, Z.; Sato, M.; Sasaki, Y.; Izumiya, K.; Kumagai, N.; Hashimoto, K. Electrochemical characterization of degradation of oxygen evolution anode for seawater electrolysis. Electrochim. Acta 2014, 116, 152–157.
- Fujimura, K.; Matsui, T.; Habazaki, H.; Kawashima, A.; Kumagai, N.; Hashimoto, K. The durability of manganese–molybdenum oxide anodes for oxygen evolution in seawater electrolysis. Electrochim. Acta 2000, 45, 2297–2303.
- Yu, J.; He, Q.; Yang, G.; Zhou, W.; Shao, Z.; Ni, M. Recent Advances and Prospective in Ruthenium-Based Materials for Electrochemical Water Splitting. ACS Catal. 2019, 9, 9973–10011.
- Exner, K.S. Beyond thermodynamic-based material-screening concepts: Kinetic scaling relations exemplified by the chlorine evolution reaction over transition-metal oxides. Electrochim. Acta 2020, 334, 135555.
- Trasatti, S. Electrocatalysis in the anodic evolution of oxygen and chlorine. Electrochim. Acta 1984, 29, 1503–1512.
- Exner, K.S. Design criteria for the competing chlorine and oxygen evolution reactions: Avoid the OCl adsorbate to enhance chlorine selectivity. Phys. Chem. Chem. Phys. 2020, 22, 22451–22458.
- Keane, T.P.; Nocera, D.G. Selective Production of Oxygen from Seawater by Oxidic Metallate Catalysts. ACS Omega 2019, 4, 12860–12864.

