Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Zaida Almeida | + 6424 word(s) | 6424 | 2021-11-05 05:17:24 |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Almeida, Z.; Brito, R. Structure and Aggregation Mechanisms in Amyloids. Encyclopedia. Available online: https://encyclopedia.pub/entry/17529 (accessed on 23 July 2026).
Almeida Z, Brito R. Structure and Aggregation Mechanisms in Amyloids. Encyclopedia. Available at: https://encyclopedia.pub/entry/17529. Accessed July 23, 2026.
Almeida, Zaida, Rui Brito. "Structure and Aggregation Mechanisms in Amyloids" Encyclopedia, https://encyclopedia.pub/entry/17529 (accessed July 23, 2026).
Almeida, Z., & Brito, R. (2021, December 23). Structure and Aggregation Mechanisms in Amyloids. In Encyclopedia. https://encyclopedia.pub/entry/17529
Almeida, Zaida and Rui Brito. "Structure and Aggregation Mechanisms in Amyloids." Encyclopedia. Web. 23 December, 2021.
Copy Citation
Functional amyloids can be found in bacteria, unicellular eukaryotes, fungi, plants, insects and vertebrates, playing roles as diverse as surface protection and modification, mediation of pathogen-host interactions, pigment biosynthesis, homeostasis control, hormone storage and release, signal transduction, among others. The aggregation of a polypeptide chain into amyloid fibrils and their accumulation and deposition into insoluble plaques and intracellular inclusions is the hallmark of several misfolding diseases known as amyloidoses. Alzheimer′s, Parkinson′s and Huntington’s diseases are some of the approximately 50 amyloid diseases described to date.
misfolding diseases
amyloidosis
oligomers
aggregates
aggregation
aggregation mechanisms
steric zipper
amyloid fibrils
amyloid structure
amyloid dyes
1. Background
The conversion of normally soluble proteins into insoluble and highly stable amyloid deposits has become the focus of attention by researchers from diverse scientific fields, from physics to chemistry, from biology to medicine. This interest results from the recognition that many of the diseases related with amyloid formation are amongst the most common and debilitating disorders of the modern era [1][2]. Many of these disorders are strongly associated with ageing, such as Alzheimer’s disease (AD) and wild-type TTR amyloidosis (ATTRwt), formerly known as senile systemic amyloidosis (SSA). Alzheimer’s disease affects approximately 50 million people worldwide and it is estimated that this number will exceed 150 million in 2050 [3]. Wild-type TTR amyloidosis affects more than 25% of people over 80 years old, and the disease is becoming more common due to the fast growth of the average age of world population and the increased awareness of medical doctors and positive diagnosis of the disease [4][5]. Presently, there are approximately 50 known amyloid disorders generally referred as protein conformational diseases, misfolding diseases or amyloidosis, with a multitude of distinct symptoms, which are associated with the misfolding and aggregation of several peptides or normally soluble and functional proteins. Table 1 lists some of the amyloid diseases and associated proteins that have been identified to date [1][2]. In the case of local amyloidosis, the amyloid deposits are observed in the organ/tissue where the precursor protein is synthesized; whereas in systemic amyloidosis, the deposition of aggregates and fibrils occurs at locations different than the sites where the precursor protein is expressed [6][7].
| Disease | Precursor Protein | Polypeptide Length (n° of Residues) | Structural Organization of Precursor |
|---|---|---|---|
| Neurodegenerative Diseases | |||
| Alzheimer’s disease | Amyloid-β variants | 37–44 | IDP |
| Spongiform encephalopathies | Prion protein or its fragments | 208 | IDP and α-helical |
| Parkinson’s disease | α-synuclein | 140 | IDP |
| Frontotemporal dementia with Parkinsonism | Tau | 352–441 | IDP |
| Amyotrophic lateral sclerosis | Superoxide dismutase 1 | 153 | β-sheet |
| Huntington’s disease | Huntingtin with polyQ expansion | 3144 | Mostly IDP |
| Neuroferritinopathy | Ferritin | 175 or 183 | α-helical |
| Familial British dementia | ABri | 34 | IDP |
| Familial Danish dementia | ADan | 34 | IDP |
| Familial amyloid polyneuropathy | Transthyretin variants | 127 | β-sheet |
| Non-Neuropathic Systemic Amyloidosis | |||
| Amyloid light chain amyloidosis | Immunoglobulin light chains or its fragments | ~90 | β-sheet |
| Amyloid heavy chain amyloidosis | Immunoglobulin heavy chains or its fragments | ~220 | β-sheet |
| Amyloid A amyloidosis | Serum amyloid A protein fragments | 45–104 | α-helical and unknown fold |
| Familial Mediterranean fever | Serum amyloid A protein fragments | 45–104 | α-helical and unknown fold |
| Apolipoprotein A1 amyloidosis | Apo A-1 fragments | 80–93 | IDP |
| Senile systemic amyloidosis | Wild-type transthyretin | 127 | β-sheet |
| Familial amyloid cardiomyopathy | Transthyretin variants | 127 | β-sheet |
| Haemodialysis-related amyloidosis | β2-microglobulin | 99 | β-sheet |
| Lysozyme amyloidosis | Lysozyme variants | 130 | α-helical and β-sheet |
| Finnish hereditary amyloidosis | Fragments of gelsolin variants | 53 or 71 | IDP |
| Non-Neuropathic Localized Amyloidosis | |||
| Type II diabetes | Islet amyloid polypeptide | 37 | IDP |
| Injection-localized amyloidosis | Insulin | 21 and 30 | α-helical |
| Gelatinous drop-like corneal dystrophy | Lactoferrin | 691 | α-helical and β-sheet |
| Medullary carcinoma of the thyroid | Calcitonin | 32 | IDP |
| Localized cutaneous amyloidosis | Galectin 7 | 136 | β-sheet |
| Atrial amyloidosis | Atrial natriuretic factor | 28 | IDP |
| Cataracts | γ-crystallins | variable | β-sheet |
IDP: Intrinsically disordered protein.
As the name suggests, protein misfolding is associated with the formation of an altered protein fold relative to the native structure of a protein. Misfolding can occur as a consequence of several events [8][9][10][11][12][13]: (1) mutations in the gene sequence leading to the production of a protein unable to adopt the native fold; (2) errors in the processes of transcription or translation leading to the production of modified proteins unable to properly fold; (3) failure of the chaperone machinery; (4) mistakes on post-translational modifications or intracellular trafficking of proteins; (5) structural modifications produced by environmental changes; (6) seeding and cross-seeding by pre-formed aggregates or surfaces. Additionally, protein misfolding generally leads to protein aggregation, with specific interactions occurring between intermediate molecular species that tend to form large ordered aggregates, which may evolve into amyloid fibrils which might deposit in the form of insoluble extracellular plaques or intracellular inclusions.
2. Protein Aggregation
In addition to their native structure that intimately correlates with function, proteins also coexist in other states, including disordered, partially unfolded, or multiple aggregation assemblies. Figure 1 summarizes the most relevant protein states, from a mechanistic and biological point of view.
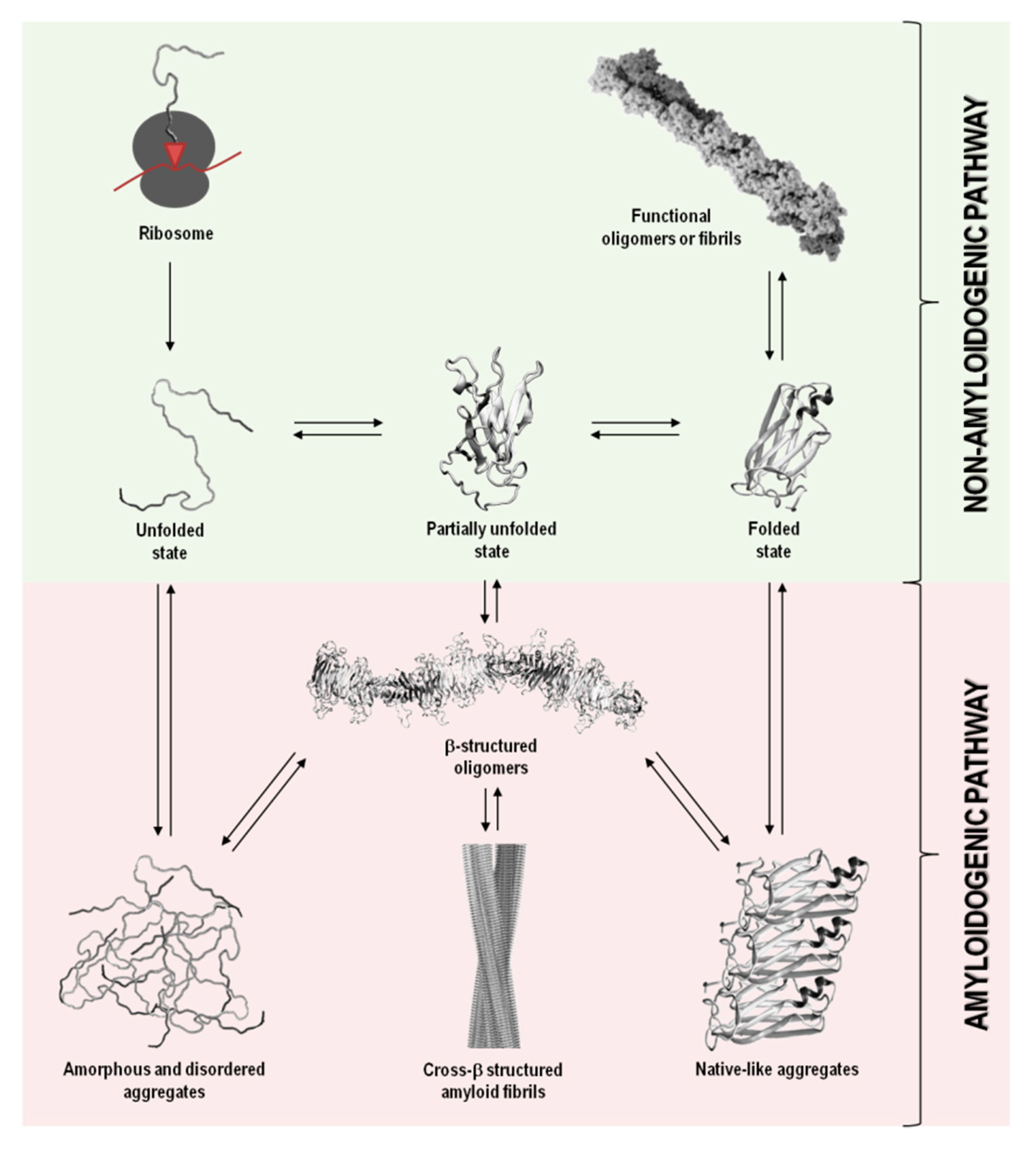
Figure 1. Schematic representation of the variety of conformational states that can be adopted by a polypeptide chain upon biosynthesis in the ribosome. The non-amyloidogenic pathway includes the formation of native sates and functional amyloids. The amyloidogenic pathway associated with pathological states can result from the formation of amorphous aggregates, amyloid aggregates and fibrils, and native-like aggregates. Adapted from reference [1].
In a functional living system, the multiple conformational states adopted by proteins involve an extremely complex series of thermodynamic equilibria and kinetic barriers, which ultimately are defined by the protein amino acid sequence. Although their intrinsic amino acid sequences and biological environments in which proteins function have co-evolved to preserve proteins in their native and soluble states (non-amyloidogenic pathway), in some conditions, proteins can interconvert into non-functional and cytotoxic protein aggregates (amyloidogenic pathway) (Figure 1) [13][14].
Protein aggregation has been shown to involve natively unfolded or intrinsically disordered systems, such as in amyloid-β (Aβ) peptide in Alzheimer’s disease, or even folded or globular proteins, such as in transthyretin (TTR) associated amyloidoses, where the misfolding process occurs through the formation of partially unfolded states (Table 1and Figure 1). In most cases, the oligomeric species formed during the amyloidogenic pathway are assemblies of monomeric units. Such aggregates can adopt highly disordered structures, well-defined fibrils with cross-β structure, or also native-like conformations, when originated from unfolded, partially unfolded, or folded monomeric states, respectively (Figure 1). All these types of aggregates have connections with amyloid disorders as they accumulate in well-characterized pathological states and represent a significant manifestation of the multiplicity of mechanisms, structures and morphologies observed during protein aggregation and disease progression (Table 1) [15].
Although the term “amyloid” is often associated with amyloid diseases, the amyloid state can also be present in functional biological processes and contribute to normal cell and tissue physiology (Figure 1). Functional amyloids can be found in bacteria, unicellular eukaryotes, fungi, plants, insects and vertebrates, playing roles as diverse as surface protection and modification, mediation of pathogen-host interactions, pigment biosynthesis, homeostasis control, hormone storage and release, signal transduction, among others [16][17][18][19].
3. Amyloid Fibrils
In 1854, Rudolph Virchow found a macroscopic tissue abnormality that exhibited a positive iodine staining reaction, suggesting the presence of starch or amylin like material (from amylum in Latin and amylon in Greek). He then suggested that the abnormal deposits could be cellulose in origin and coined the name “amyloid”. Friedrich and Kekule, in 1859, showed that the deposits were actually protein aggregates, but the designation remained and is still used today [20]. Thus, abnormal accumulations of insoluble highly ordered and fibrillar protein structures in tissues and organs, such as in “misfolding diseases” (Table 1), are known as amyloid fibrils (Figure 1). No sequence, structural or functional correlations are apparent between many of the proteins that display the ability to form amyloid fibrils. The number of amino acids is diverse ranging, for example, from 37 to 44 in Aβ peptide to 691 in lactoferrin (Table 1). Despite these dissimilarities, amyloid fibrils from different protein precursors share many common structural features, namely, the formation of long, unbranched filaments with a cross-β structural motif [21][22][23].
3.1. The Tinctorial Properties of Amyloid Fibrils
From the early days in amyloid research, molecular probes have been used in order to both characterize the mechanisms of amyloid fibril formation, and detect the presence of amyloid in tissues and samples, taking advantage of the distinctive structural features and tinctorial properties of the amyloid material. These molecular probes display changes in spectroscopic properties upon binding to amyloid fibrils. Usually, they are aromatic heterocyclic compounds, some of which toxic [24]. Congo red (CR) and thioflavin-T (ThT), shown in Figure 2, are the most commonly used dyes to identify amyloid material and study amyloid aggregation [25][26][27].
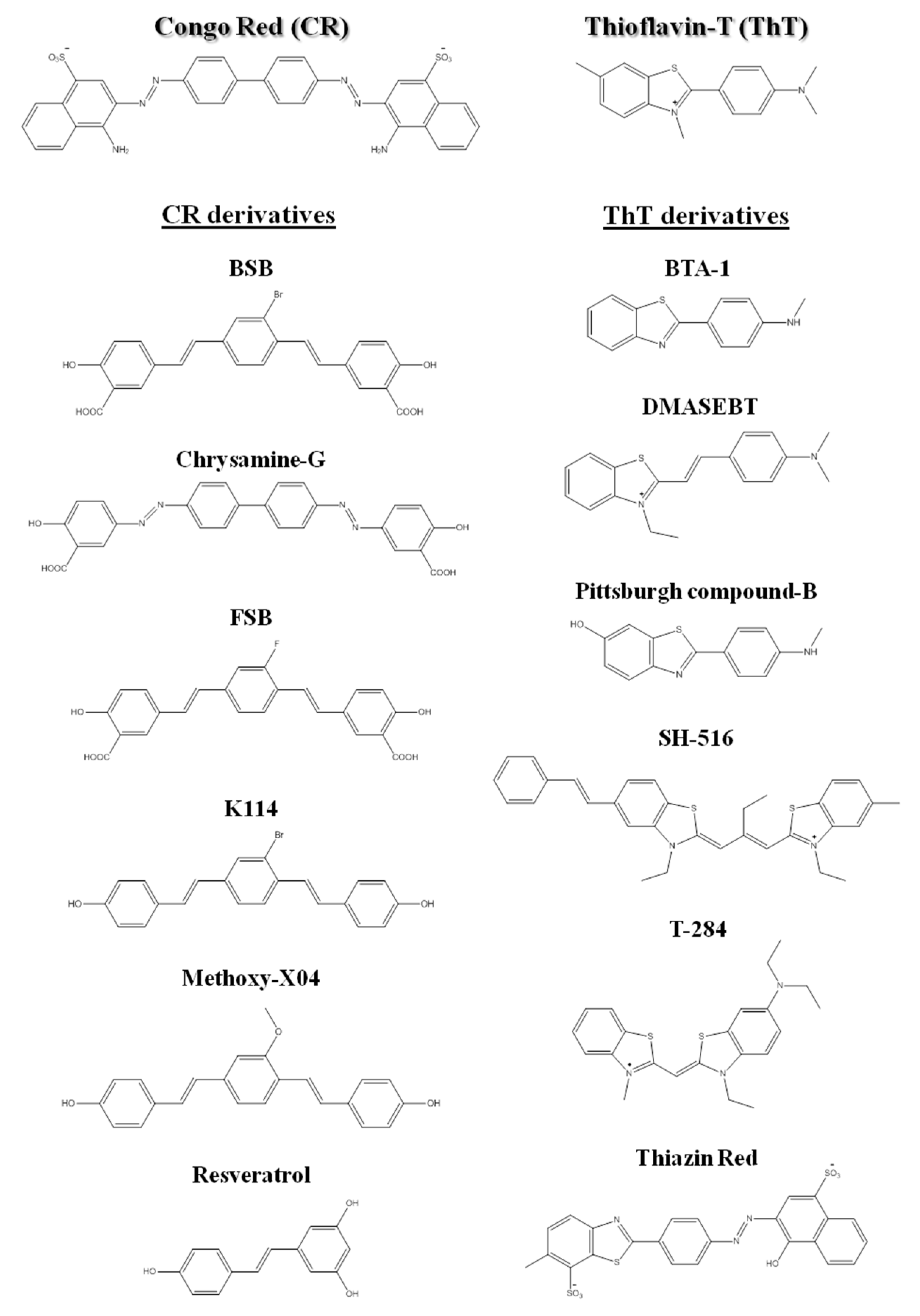
Figure 2. Chemical structures of Congo red, thioflavin-T, and some of their derivatives used in amyloid fibril detection.
Since the beginning of the 1920s, Congo red has been used for detection of amyloid fibrils. Upon binding to amyloid fibrils, CR displays green birefringence under polarized light, which is associated with a characteristic red shift in the absorbance maximum (from 490 to 512 nm), and presence of a characteristic shoulder peak at approximately 540 nm [[28]]. CR is the most commonly applied dye in the identification of amyloid in ex vivo tissue slices, using mainly polarization microscopy, but also light microscopy [[29]] and fluorescence microscopy [[30]]. However, compared to ThT, CR is less sensitive in the detection of amyloid fibrils [[31]]. In addition, CR interferes with the aggregation mechanisms and is not appropriate for in situ detection, since it is known to inhibit or to enhance amyloid fibril formation depending on the protein [32][33][34][35][36][37].
More recently, thioflavin-T became a standard dye for amyloid detection showing fluorescence enhancement upon interaction with amyloid deposits in tissue sections [38]. ThT fluorescence emission increases upon binding to amyloid fibrils, with excitation at 450 nm and emission maxima at 482 nm, as opposed to excitation at 385 nm and emission maxima at 445 nm for the free dye [31].
Besides ThT and CR, other compounds have been also used for the detection of amyloid fibrils [39][40][41][42]. The search for novel, more sensitive and blood–brain barrier crossing dyes for detecting the initial stages and more toxic oligomeric species involved in fibril formation in vitro, ex vivo and in vivo is still under way [43][44][45]. Nevertheless, several related structures or derivatives of both CR [46][47][48][49] and ThT [50][51][52][53] have been shown to be quite helpful for both in vitro and ex vivo identification of amyloid fibrils (Figure 2).
The binding mode of the molecular probes to amyloid fibrils is not fully understood. In the case of ThT and CR, a large number of studies examined binding modes and suggested the existence of a binding interface parallel to the long axis of the fibril [54][55].
Features as fibrillar morphology, cross-β structure, and characteristic tinctorial properties are nowadays accepted as benchmarks for defining “amyloid materials”, and any given protein aggregates need to display most of them to be classified as such. In a recent recommendation, the International Society of Amyloidosis (ISA) nomenclature committee suggests the following classification for amyloids based on five main classes, in order to distinguish different natural and synthetic amyloid-like materials: (1) in vivo and ex vivo disease-related fibrils; (2) in vivo and ex vivo functional fibrils; (3) recombinant fibrils of disease-related proteins or functional amyloid proteins; (4) fibrils from synthetic or non-disease-related peptides; and (5) fibrils from hydrogels that produce the cross-β diffraction pattern [56].
3.2. Structure of Amyloid Fibrils at the Subunit Level
Since the cross-β motif was identified [57], many studies have been performed to characterize the structure of amyloid fibrils, using different experimental techniques. The combination of all these data has contributed to reveal the structure of amyloid fibrils on a multi-scale basis and to show how individual protein subunits can form cross-β structures.
The structural feature which all amyloid fibrils share is the cross-β motif, characterized by extended β-sheets with individual β-strands arranged in an orientation perpendicularly to the fibril main axis. Amyloid fibrils are unbranched structures with diameters ranging from 2 to 20 nm and usually present several micrometers in length [21][58][59]. The existence of a repeating cross-β structure was first demonstrated in the late 1960s by X-ray fiber diffraction studies [60][61], and later synchrotron X-ray diffraction studies have shown that amyloid fibrils from different amyloid proteins exhibit the same cross-β diffraction pattern [62]. Characteristic amyloid fibril diffraction patterns show a meridional reflection at 4.7–4.8 Å corresponding to the hydrogen bonding distances found between paired carbonyl and amide groups in adjacent β-strands, and an equatorial reflection at 6–11 Å corresponding to the distance observed between stacked β-sheets [62].
Amyloid fibrils from different origins contain a common cross-β spine formed by a pair of β-sheets with the facing side chains of the two sheets interdigitated in a steric zipper [63]. Steric zippers are thus short peptide segments which provide the structural basis for the hierarchical assembly of an amyloid fibril [63][64]. In this sense, stacks of steric zippers are needed to form the cross-β spine of the amyloid protofilament (Figure 3) [65], which constitutes the basic unit of the mature fibril, whereas the rest of the polypeptide chain assumes either native-like or random coil conformation in a peripheral position to the spine [63][66].
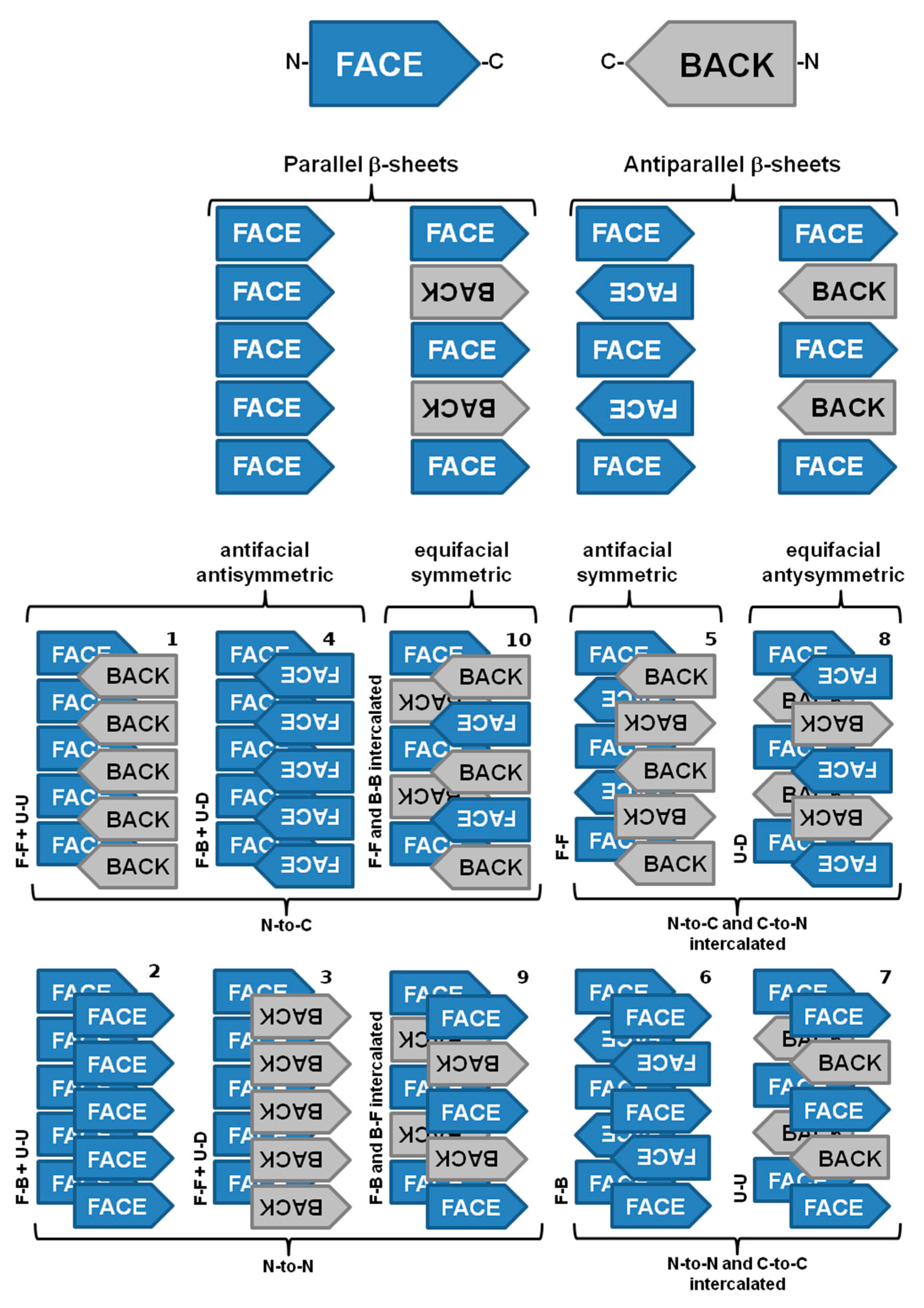
Figure 3. The ten possible amyloid symmetry classes of homo-steric zipper amyloid spines. Legend: face (F), back (B), up (U), down (D), N-terminal and C-terminal. Adapted from reference [65].
Nevertheless, significant variations may be found among the structural arrangement of steric zippers. Assembly of the strands in a sheet produces four patterns of sheet symmetry (parallel versus antiparallel, and antifacial versus equifacial). These four β-sheet patterns are each capable of self-pairing to form cross-β spines. Thus, in total, ten distinct symmetry classes of steric zippers can be enumerated. These classes are distinguishable by the particular sheet pattern and by the symmetry operation that relates the pair of sheets (Figure 3) [65].
Mature fibrils also reveal a periodic structure due to the twist of multiple protofilaments around each other. The helical twist of the protofilaments can give rise to a discernible overall helicity of the mature amyloid fibril. Although the basic structural arrangement of the cross-β structure is conserved in different fibrils, there are different possibilities of how protofilaments can pack into the three-dimensional fibril structure, giving rise to several distinct amyloid fibril morphologies. It is possible to distinguish amyloid fibril polymorphism based on the degree of fibril twisting, the number of protofilaments per fibril, and the diameter or weight per length of the fibrils [22][67][68]. In addition to amino acid composition, the fibril morphology is determined by environmental factors, such as pH, temperature, mechanic agitation, shear force, ionic strength or presence of other co-solutes and co-solvents [22][69].
Amyloidogenic regions or short segments forming steric zipper spines in amyloid fibrils are possible to identify/predict by submitting a protein sequence to specific algorithms and web services. Table 2 summarizes the computational tools available online and reported in the literature to predict propensities or incidence levels of a given amino acid sequence to form amyloid fibrils.
Table 2. Available online methods to predict amyloidogenic regions in amino acid sequences.
3.3. The Cross-α Amyloid-Like Fibril
Cross-β motifs and amyloid fibrils can be formed both by β-sheet rich and α-helical proteins. Alpha-helical conformations have also been associated to amyloid fibril formation. Certain peptides and proteins that present well defined α-helical structures undergo conformational changes into β-pleated structures that in turn aggregate into amyloid fibrils [103][104][105][106][107], while other peptides or proteins maintain its α-helical conformation during the aggregation cascade, forming “cross-α” structures. Cross-α structures are also formed by elongated unbranched fibrils, and display the ability to bind thioflavin-T with the characteristic enhanced fluorescence emission. These fibrils result from the stacking of α-helices perpendicular to the fibril axis [108]. The cross-α structure, although rare, was found in the functional amyloid phenol soluble modulin α3 (PSMα3) [108], in the yeast prion Ure2p [109], in de novo-designed amphiphilic peptides [110][111][112], and among proteins of multiple tandem copies of a helix–loop–helix unit [113].
3.4. Hetero-Amyloid Fibrils
The terms “hetero-amyloid” or “hetero-fibrils” relate to amyloid fibrils structures formed by two different β-sheets along a compact hydrophobic interface, featuring an unusual ladder of sequential stacking. The hetero-fibril is stabilized by hydrophobic packing and enriched with interactions along the fibrillar axis, such as hydrogen bonds between neighboring β-strands, analogous to homo-amyloids. This type of amyloid structure was found in the interaction between the M45 protein with other proteins, namely the receptor-interacting protein kinase 1 and 3 (RIPK1 and RIPK3), and the Z-DNA binding protein-1 (ZBP1) [114]; as well as in the RIPK1-RIPK3 necrosome human signaling complex [115].
In addition, it has also been observed that some amyloidogenic proteins may support cross-seeding by amyloid aggregates of different proteins, building a link between different amyloid forms. Heterologous cross-seeding has been reported between amyloid-β peptide (Aβ) and islet amyloid polypeptide [116][117], Aβ and prion protein (PrP) [118], and Aβ and α-synuclein [119]. Furthermore, Aβ fibrils also induce the formation of tau neurofibrillary tangles in vivo [120], and Aβ aggregates promote tau aggregation in vitro [121]. Heterologous cross-seeding was also found when tau K18 interacts with the C-terminus of α-synuclein [122], when functional amyloids curli and Sup35 induce amyloid protein A amyloidosis [123], when PrPs promote Sup35 aggregation [124], and when the in vitro aggregation of CsgA is seeded by CsgB fibrils [125]. Moreover, the observation of heterologous cross-seeding cases suggests that interactions between different amyloidogenic proteins might enhance the onset of certain types of amyloidoses.
3.5. The Major Differences between the Techniques that Inform on Amyloid Structure
Recent progresses in instrumentation have improved quite significantly the ability to characterize the structure of complex biological molecules and macromolecular complexes. The knowledge of the three-dimensional structure of amyloid fibrils from different origins provides important insights into the mechanisms of amyloid formation, and thereby helps in the rational design of novel therapeutic agents. The combination of structural biology data from several approaches and experimental techniques provides new insights on how individual protein subunits form fibrillar structures, at an atomic level of resolution. In the past decades, many structural biology studies have been performed based on X-ray fiber diffraction [126], X-ray crystallography [23], solid state NMR (ssNMR) [127], and cryo-electron microscopy (cryo-EM) [128], to mention just a few. Table 3 reports the main differences between these powerful techniques. However, is important to mention that no method is completely effective by itself, since all of them offer unique advantages and limitations.
Table 3. The pros and cons of the main methods that inform on amyloid structure at atomic level resolution.
| Technique | Advances | Advantages | Disadvantages | References |
|---|---|---|---|---|
| X-ray and electron diffraction | 1. Discovery of short protein segments that can themselves form amyloid fibrils and closely related crystals; 2. Development of synchrotron X-ray microbeams sufficiently focused and intense to determine a structure from a single crystal. |
1. May yield atomic resolution; 2. Is not limited by the molecular weight of the specimen. |
1. Well-ordered microcrystals needed; 2. The fibrils formed by some segments may represent the spines of polymorphs of full fibrils, but others may not; 3. The crystallized segment is only a few residues in length, thus nothing is revealed about the fibril structure outside the spine; 4. The steric zippers structures only show homo-steric zippers. |
[23][60][62][126] |
| ssNMR | 1. Innovations in high-field magnets, pulse sequences, high-resolution multi-channel magic-angle spinning (MAS) probes, ultrafast MAS, isotopic labeling schemes, use of quadrupolar nuclei as spectroscopic probes and solid-state dynamic nuclear polarization (DNP). | 1. No need for crystals; 2. Structural information obtained on: identity of residues, recognition of parallel versus antiparallel β-sheets, register of strands within a sheet, and inter-residue contacts of amino acid side chains; 3. ssNMR-determined models show the overall conformation of the well-ordered portion of the chain around the protofilament spine; 4. Can be used to determine dihedral angles and inter-atom distances in the fibril subunits. |
1. Amyloid-forming proteins are expressed recombinantly from media containing isotopically labeled amino acids; 2. Reliability of molecular models is highly dependent on the number of experimental constraints that have been collected; 3. The relative positions of atoms are not as accurately determined as in an atomic-resolution crystal structure; 4. The sensitivity of the experiments and spectral resolution decrease with the increase in molecular weight. |
[127][129][130] |
| cryo-EM | 1. Introduction of high-field microscopes; 2. New generation of direct detectors record the incident electrons in a thin, sensitive layer so that the signal is not scattered into surrounding pixels resulting in an improvement in image processing. |
1. Near atomic-resolution structures of large molecular complexes without the need for crystals; 2. May yield the overall fibril structure: the number of protofilaments; the degree of twist; and, depending on the number of well-ordered specimens, information on the atomic structure of the fibril. |
1. Due to a lack of contrast, images often have a very low signal-to-noise ratio, requiring highly advanced detection hardware and image processing; 2. Sample preparation can be difficult, not only to optimize thickness, but also to optimize particle distribution; 3. The most advanced cryo-EM equipment is very expensive. |
[131][132][133] |
4. Toxic Species in Amyloid Diseases
The pathogenic or the most toxic species in amyloidosis can result from extracellular amyloid fibrillar deposits affecting organ integrity [134][135][136][137], as in the case of cardiac amyloid in transthyretin amyloidosis, and/or soluble protein oligomers that can either populate during the process of amyloid fibril formation or be released by mature deposits causing direct cellular dysfunction [137], as in the case of several neurodegenerative amyloid diseases.
Although the β-sheet structure present in amyloid oligomers is a favorable structural component, it is not an important prerequisite for toxicity [138]. The structural determinants by which these misfolded oligomers cause cellular damage may be related with the exposure of hydrophobic groups on the oligomer surface [139][140] and with the small size of the oligomers with high diffusion coefficient [140][141]. Thus, the toxicity of amyloidogenic oligomers is likely to be a result of their intrinsic misfolded nature and aggregation propensity [137][142]. Such structural properties will cause them to engage in a multitude of abnormal interactions with a range of cellular components, such as phospholipid bilayers, soluble peptides and proteins, protein receptors, RNAs, and cellular metabolites [139][143][144][145][146][147] where some or all of them have the potential to cause cell damage and ultimately cell death. In fact, it is known that several amyloid neurodegenerative diseases have an important inflammatory component which may be the result of the natural response against the “unknown” molecular species formed during the amyloidogenic cascade or against by-products of their action.
5. Kinetics and Thermodynamics of Amyloid Fibril Formation
The aggregation mechanisms in amyloid systems are prone to multiple pathways, depending on the ensemble of co-existing amyloidogenic conformations and environmental factors. Thus, several aggregation mechanisms and multiple pathways have been described depending on protein sequence, conformational states adopted by the amyloidogenic monomer and experimental conditions (for instance, temperature, pH, protein concentration, and solvent effects). The aggregation processes take place over a wide time range, spanning several orders of magnitude, with conformational changes occurring in the milliseconds and formation of particles observable with the naked eye in days, weeks or months. Elucidation of the mechanisms of amyloid formation and characterization of the most relevant molecular species involved are crucial to devise new rational therapeutic strategies against amyloid diseases. A brief overview of the most common amyloid formation mechanisms is presented in Figure 4, and a summary of the events and conditions that may trigger protein aggregation is shown in Table 4.
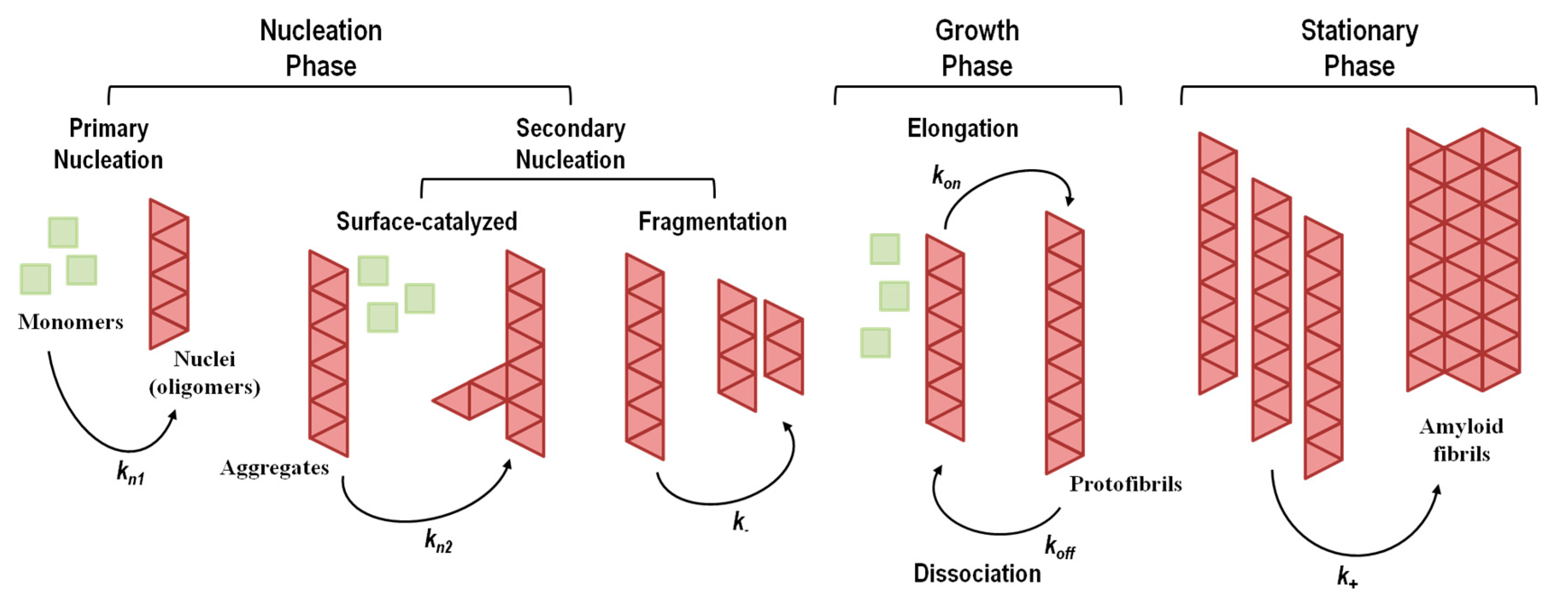
Figure 4. Representative general model for amyloid fibril formation by nucleation-dependent mechanisms (including primary and secondary nucleation) and nucleation-independent mechanisms (absence of nucleation). kn1, kn2, k−, kon, koff, and k+ represent rate constants. The stationary phase involves the assembly of protofibrils into mature amyloid fibrils with different morphological structures and a high level of polymorphism. Adapted from reference [148].
Table 4. Early events that may lead to amyloid fibril formation.
| Nature of Monomeric Species | Description | References |
| Native monomer |
Normally folded proteins may retain a substantial tendency to aggregate through direct assembly of monomers in their native state when the native state exposes complementary surfaces. | [153][154][155][156] |
| Conformationally altered monomer | The native monomer has very low propensity to associate. Partial unfolding or conformational changes of the native monomer are required, resulting in a non-native species prone to aggregate. | [157][158][159][160][161][162] |
| Chemically modified monomer | Chemical modifications (deamidation, isomerization, hydrolysis, oxidation, photolysis, etc.) may cause conformational changes in native monomers, leading to species with high propensity to aggregate. | [163][164][165][166][167][168][169][170][171][172][173][174] |
| Nature of Aggregation Interfaces | Description | References |
| Gas-liquid interface |
Hydrophobic–hydrophilic interfaces may induce aggregation reactions. | [175][176][177][178][179][180][181] |
| Mechanical stress (agitation, stirring, pumping, or shaking) has been associated with cavitation which generates air bubbles and, consequently, the formation of an air-water interface which facilitates protein denaturation and aggregation. | [176][182][183][184][185][186][187][188][189] | |
| The use of beads during agitation accelerates the aggregation process by enhancing cavitation. | [190] | |
| Solid-liquid interface |
Solid-liquid interfaces may facilitate monomer encounters and initial monomer to monomer association and later further aggregation. | |
| In vitro, interaction with glass, silicone, graphite, polypropylene, Teflon, mica, gold, etc. might lead to protein partial unfolding and aggregation. | [181][191][192][193][194] | |
| In vitro and in vivo, flow through tubes and vessels produce shear forces that may lead to protein partial unfolding and aggregation. | [195] | |
| Freeze-thaw cycles create new ice-water interfaces which may induce protein partial unfolding and aggregation. | [189][191][196] | |
| Presence of metal ions, in particular, Cu2+ and Zn2+, may promote aggregation of protein monomers bearing metal-ion binding sites or binding residues (e.g., histidines). | [197][198][199][200][201] | |
| Monomer association at the surface of biomembranes or biomolecules may also enhance aggregation. | [202][203][204][205][206][207][208][209][210] |
5.1. Aggregation Via a Nucleation-Dependent Mechanism
The nucleation-dependent mechanism of amyloid formation (Figure 4), also known as nucleation–elongation polymerization, displays a typical sigmoidal shape curve as a function of time and consists of three consecutive steps: (1) initial lag or nucleation phase; (2) elongation, growth, polymerization, or fibrillation phase; (3) equilibrium, stationary, or saturation phase [211][212].
The nucleation phase corresponds to the assembly of transient, critical nuclei that next will act as seeding intermediates where additional monomeric subunits can latch on, driving the assembly of oligomers with cross-β structure. At this stage, the rate constants for monomer addition and dissociation are similar, making the global process of nucleation slow and the rate limiting step in fibril formation. The nucleation phase can be shortened or eliminated by the addition of pre-formed aggregates or fibrillar species, a process known as seeding [213][214][215]. In the elongation phase, monomers, nucleus and oligomers continue to interact, assembling into prefibrillar structures that rapidly grow to form ordered fibrillar structures known as protofibrils. Because this phase gives rise to more stable protofibrils, this is a faster and thermodynamically favorable process. Lastly, the saturation phase, where monomer concentration is low and approximately constant, involves the assembly of protofibrils into mature amyloid fibrils with different morphological structures and different levels of polymorphism.
The Finke–Watzky aggregation model (Scheme 1) is one of the numerous models proposed for nucleation–elongation polymerization and has been applied to more than 40 different aggregating proteins [216][217]. As shown in Scheme 1, the Finke–Watzky model consists of two simple steps: (1) nucleation and (2) growth. Due to its simplicity, this model does have some limitations, including: (1) a vast number of aggregation steps is condensed into two elementary steps; (2) the rate constants, knucleation and kgrowth, are average rate constants and independent of the size of aggregating species; (3) a higher kinetic order in [M] may be kinetically hidden in the nucleation step in particular; (4) all growing polydisperse aggregates are hidden behind the descriptor “An”; (5) the descriptor “An” can also hide processes such as fragmentation. However, the simplicity and quality of the fits obtained in many practical examples suggest that the Finke–Watzky two-step mechanism encompasses the main characteristics and it is a good general kinetic model for nucleation-growth aggregation.
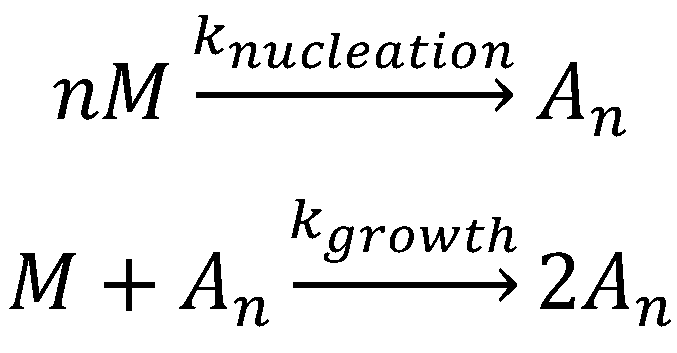
5.1.1. Primary Nucleation Mechanisms
In the amyloidogenesis cascade, primary nucleation is a critical step in a variety of oligomerization mechanisms and includes the initial formation of amyloidogenic nuclei without contributions from pre-formed oligomers. Two types of primary nucleation mechanisms have been described: homogeneous and heterogeneous. Homogeneous primary nucleation comprises monomers aggregation in bulk solution, while heterogeneous primary nucleation involves association of monomeric subunits on the surface of a different object, such as the wall of a reaction container [191][192][193][194], other proteins [202], phospholipid bilayers [203][204][205][206][207][208][209][210], or the air–water interface [175][176][177][178][179][180].
From a structural point of view, the simplest manifestation of a primary nucleation mechanism is the nucleated polymerization (NP) mechanism. In this case, amyloidogenic monomers aggregate and originate the nucleus, which further grows into amyloid protofilaments and protofibrils through an elongation process involving mostly monomer addition [219][220][221]. This is the preferential mechanism at relatively low protein concentrations favoring the presence of monomeric species in solution.
However, in several instances, it has been observed the presence of multiple conformational heterogeneous oligomers and transient intermediate species during fibril formation which NP mechanisms cannot explain. In these cases, a nucleated conformational conversion (NCC) mechanism has been proposed. NCC comprises structurally organized oligomers as intermediates which are able to subsequently conformationally transition into cross-β dominated fibrillar species. The formation of these conformationally dynamic oligomers may be favored at higher protein concentrations and they undergo a rate-limiting conformational change to form protofibrils and then amyloid fibrils [222]. This type of nucleation was observed, in particular, in the yeast prion protein (Sup35) [223], among variants of the amyloid-β peptide [224][225][226], SH3 domain [227][228][229], Ure2p yeast prion [230], polyglutamine (polyGln) peptides [231], and lysozyme [232][233].
5.1.2. Secondary Nucleation Mechanisms
Although conceptually appealing and observed in several instances, a simple homogeneous primary nucleation mechanism is not always observed [149][223][234]. Several studies have pointed out that simple homogeneous nucleation could not fit certain experimental aggregation kinetics data [235][236]. Simple homogeneous primary nucleation does not take into account other nucleation mechanisms and events, such as fibril-catalyzed secondary nucleation (a monomer-dependent process) and fibril fragmentation (a monomer-independent process) (Figure 4), both contributing to the formation of new aggregation nuclei [237][238][239][240][241]. In fibril-catalyzed secondary nucleation nucleus formation occurs on the surface of an already existing oligomer (Figure 4). No foreign surface is involved in this type of nucleation as in the case of heterogeneous primary nucleation. This nucleation mechanism appears to be highly dependent on the structural compatibility of the amyloid precursor protein [241].
Secondary nucleation has been inferred for several proteins, including amyloid-β peptides [235][242], tau protein [243], α-synuclein [244][245], islet amyloid polypeptide (IAPP) [246], insulin [247], and bovine carbonic anhydrase [248].
Amyloid fibril formation may also be seeded by the presence of pre-formed aggregates. In this case, the primary nucleation event is negligible, leading directly to the growth phase, the absence of secondary mechanisms, and the polymerization process is expected to follow a single exponential function [249]. This is a consequence of the slower rate of primary nucleation when compared with the rate of addition of monomers onto an existing fibril (growth). This seeding process has been proposed to be an important factor in the propagation of the pathogenesis in most, if not all, amyloidoses [250][251][252][253][254][255][256].
5.2. Aggregation Via a Nucleation-Independent Mechanism
The nucleation-independent mechanism of protein aggregation (Figure 4 and Scheme 2) is an isodesmic or linear polymerization mechanism and may be exemplified by the simplest possible model for the formation of spherical oligomers or linear multimers [[152]]. This model is characterized by an infinite number of steps with identical rate constants (k) independent of the size of the aggregate (Scheme 2), resulting in an exponential polymerization curve with the absence of a lag phase. Once aggregation starts, the process undergoes downhill-polymerization. In this case, aggregation proceeds through a sequence of multiple energetically favorable steps, where the successive addition of amyloidogenic monomers to the growing aggregate is energetically favorable without the need of a multimeric nucleus.
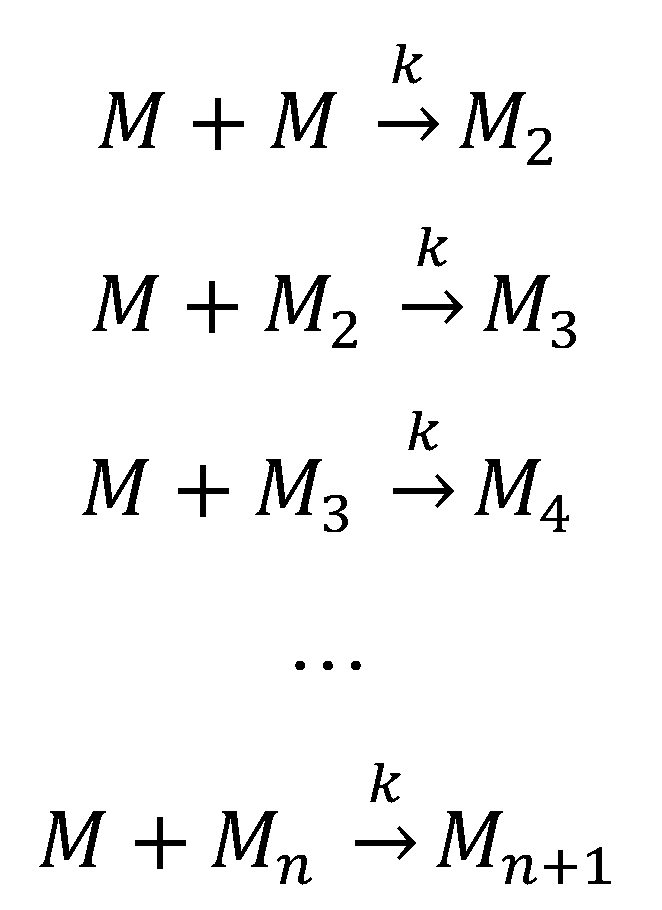
Generally, seeding does not increase the aggregation rate in a downhill-polymerization process. However, this model disregards other aggregation processes that can change the number, size and shape of oligomeric species. For these reasons, this model sometimes predicts incorrect length distributions of amyloid fibrils at equilibrium. Nevertheless, this kinetic model has been used to investigate the effect of mutations on the rate of amyloid fibril formation [259][260][261].
This aggregation mechanism was observed in transthyretin [262][263], among variants of the amyloid-β peptide [219][264][265], and also in the four-repeat domain of tau (Tau4RD) [266], β2-microglobulin [152], human and bovine serum albumins [267][268], HypF-N [269], FF domain [270], human muscle acylphosphatase [271], apolipoprotein C-II [272][273], and several SH3 domains [228][259][274][275][276].
5.3. The Energy Landscape View of Protein Aggregation
The protein folding energy landscape available to each polypeptide chain includes a wide range of different conformational states and a multitude of pathways en route to the folded state. The energy landscape in the case of short polypeptide sequences tends to be a smooth funnel-shaped surface where the polypeptide chain folds quickly towards a single folded state [277]. On the other hand, larger proteins have rougher energy landscapes, with local minima and a population of intermediate states that eventually interconvert to the low energy folded state (Figure 5) [278][279].
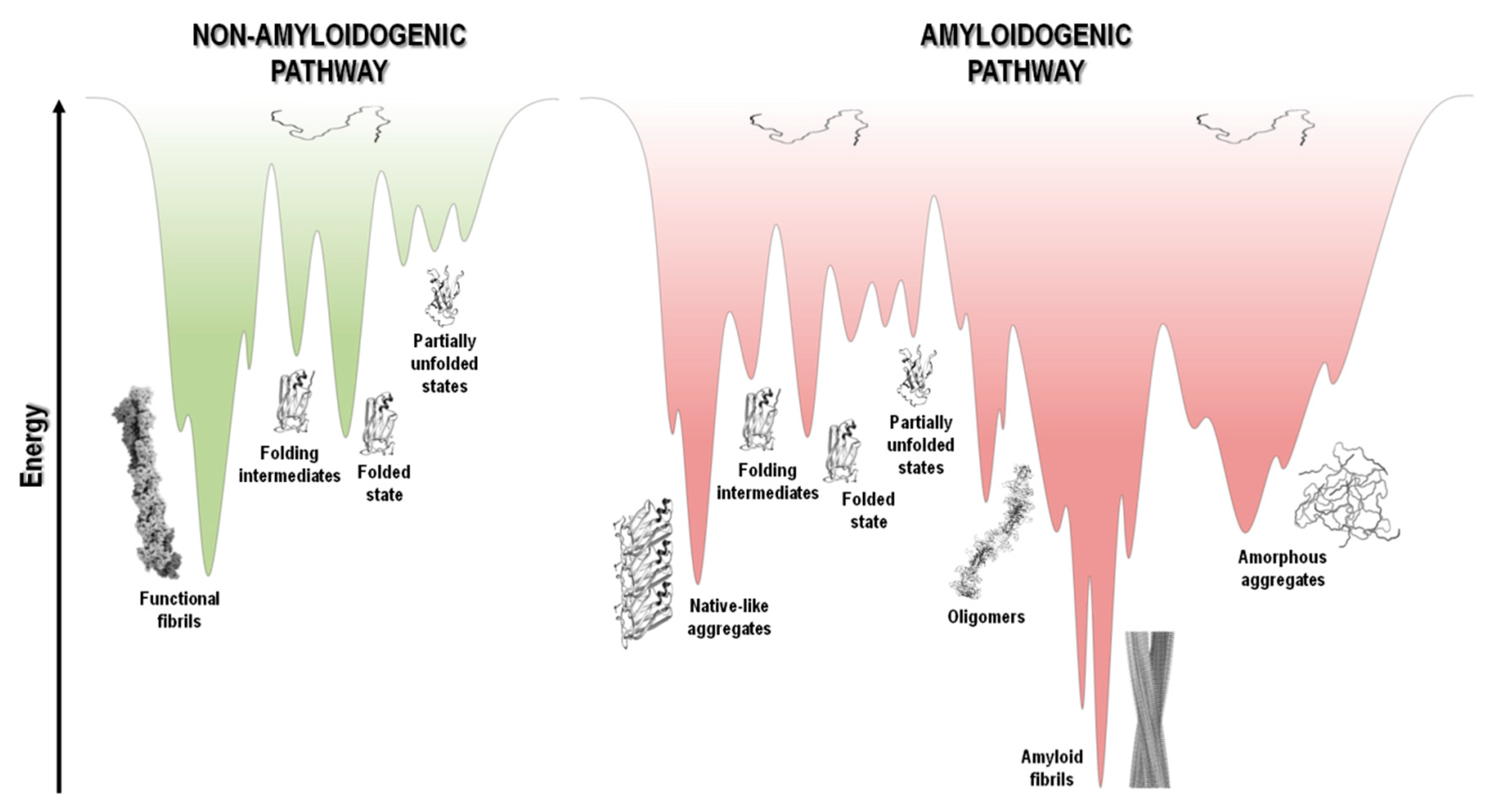
Figure 5. Schematic representation of funnel-shaped energy landscapes for protein folding (non-amyloidogenic pathway, green) and aggregation (amyloidogenic pathway, red). The surface exhibits the roughness of the protein energy landscape showing the possible conformational states adopted by the polypeptide chain. Unfolded, partially unfolded and folded species may be implicated in the aggregation landscape, as long as they are prone to establish intermolecular interactions and aggregate, thereby resulting in the formation of amorphous aggregates, amyloid fibrils, and native-like aggregates, respectively. Adapted from references [1][279][280].
At the top of the funnel, as depicted in Figure 5, the unfolded state of the polypeptide chain has high Gibbs free energy and high conformational entropy. Upon polypeptide chain folding, the number of conformational states and thus the conformational entropy decreases. Concurrently, the hydrophobic collapse and the increase in the number of intramolecular contacts leads to a decrease in free energy toward the native state occupying the global free energy minimum, yielding the necessary conformational stability of the folded state. However, changes in amino acid sequence, and/or chemical or biological environment, including changes in pH, temperature, ionic strength, pressure, agitation, shear forces, interaction with surfaces and many other factors, may tip the energetic balance towards a different free energy minimum. This is highlighted in Figure 5, where in parallel with the common folding funnel for a protein it is also depicted an aggregation funnel [280][281][282]. Regarding the protein aggregation process, the funnel-shaped free energy surface is potentially rougher and more complex, since the energy landscape encodes not only the relative stability of unfolded states, partially unfolded states, and folded states, but also the relative stability of amorphous aggregates, β-sheet-rich amyloid fibrils, and native-like aggregates (Figure 1 and Figure 5).
Protein aggregation involves several processes that are interconnected, such as folding, unfolding and partial folding/unfolding, conformational changes, formation of intermolecular interactions, and fibril nucleation, elongation and stationary phases (Figure 1 and Figure 4) [237][238][283][284][285]. There is well documented evidence that protein aggregation states may be formed not only by amyloidogenic intermediates but also by denatured and native states, with polypeptide chains establishing critical contacts with neighboring molecules through intermolecular interactions. Aggregated states are in general thermodynamically and kinetically favorable and it is a fine balance of forces that tip the processes towards a native soluble state or any type of aggregated state. It seems that most polypeptide chains under the right “stress” conditions tend to form extended β-sheet structures and thus amyloid aggregates [14][284][286][287]. The energy landscape of protein systems forming large aggregates is described by numerous peaks corresponding to different conformational states, which is the case of amyloids due to the heterogeneity of fibrillar morphology. Even under the same experimental conditions, a large number of polymorphic fibrils with distinct morphologies might be formed at the same time, emphasizing the complexity and multiplicity of the aggregation pathways [67][288]. The energy minimum of mature fibrils is deeper and sharper than the native state of a given protein (Figure 5), as suggested by the high stability of the fibrillar state [286][289][290].
Under specified conditions, a given polypeptide chain has its own folding and aggregation surface funnel. Each point on this surface expresses a specific and unique conformation of the protein. The profile of the energy landscape is affected by enthalpic contributions due to interactions between amino acids residues, and enthalpic and entropic contributions due to the interaction with the aqueous environment, as well as entropic contributions due to changes in conformational freedom of the polypeptide chain. The driving forces towards the low free energy state for both protein folding and aggregation are mainly hydrophobic in nature, with additional contributions from electrostatic and polar interactions, as well as hydrogen bonds [14][280][281][287]. In the case of amyloid fibrils, the cross-β motif conformation is stabilized essentially by polar interactions due to intermolecular hydrogen bonds, and intermediate aggregated species are formed by intermolecular hydrophobic and electrostatic interactions [284][291].
In the late 1990s [292] amyloid-like aggregates or fibrils were found to be formed in vitro under specific experimental conditions by proteins entirely unrelated to well-established amyloid diseases [293]. As we previously mentioned, under the right conditions, many if not most polypeptide chains may form amyloid. The term “amylome” was thus coined to describe all the proteins that can form amyloid-like fibrils [294]. There are authors that consider that the fibrillar amyloid state represents a standard state that every polypeptide chain can adopt under appropriate conditions, and that this state is the thermodynamic ground state. In this sense, the amyloid fibrillar conformation would be the universal global free-energy minimum of any polypeptide chain [295].
6. Conclusions
With the progresses being made in medical research and drug discovery, there is an optimistic view that protein misfolding diseases will become successfully diagnosed, prevented and treated. At the moment, the research into the “amyloid problem” is reaching a turning point, since the structures and properties of different amyloidogenic species involved in the amyloid cascade are finally being identified and, in some cases, structurally characterized. The improvement in resolution of molecular structures, with the contribution from X-ray, ssNMR and cryo-electron microscopy, has allowed a better understanding of how amyloid polymorphism may relate with different manifestations of amyloid diseases, and how different amyloid structures influence cellular function and may be influenced by tissue environment. This seems to be the perfect time to finally correlate protein aggregation mechanisms, structure of amyloid species, and how they impact the onset, severity and progression of different misfolding diseases. In turn, this is also the opportunity to set up therapeutic solutions based on rational approaches, and thus provide new hope to those many affected by amyloid diseases.
References
- Fabrizio Chiti; Christopher M. Dobson; Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annual Review of Biochemistry 2017, 86, 27-68, 10.1146/annurev-biochem-061516-045115.
- Matthew G. Iadanza; Matthew P. Jackson; Eric W. Hewitt; Neil A. Ranson; Sheena E. Radford; A new era for understanding amyloid structures and disease. Nature Reviews Molecular Cell Biology 2018, 19, 755-773, 10.1038/s41580-018-0060-8.
- Patterson, C. World Alzheimer Report 2018. The State of the Art of Dementia Research: New Frontiers; Alzheimer’s Disease International (ADI): London, UK, 2018; pp. 6–7.
- P. Westermark; K. Sletten; B. Johansson; G. G. Cornwell; Fibril in senile systemic amyloidosis is derived from normal transthyretin.. Proceedings of the National Academy of Sciences 1990, 87, 2843-2845, 10.1073/pnas.87.7.2843.
- Sanjay M. Banypersad; James C. Moon; Carol Whelan; Philip N. Hawkins; Ashutosh D. Wechalekar; Updates in Cardiac Amyloidosis: A Review. Journal of the American Heart Association 2012, 1, e000364, 10.1161/jaha.111.000364.
- Sipe, J.D. Amyloidosis. Annu. Rev. Biochem. 1992, 61, 947–975.
- Luis M. Blancas-Mejía; Marina Ramirez-Alvarado; Systemic Amyloidoses. Annual Review of Biochemistry 2013, 82, 745-774, 10.1146/annurev-biochem-072611-130030.
- Alexandre Quintas; Maria João Saraiva; Rui M.M Brito; The amyloidogenic potential of transthyretin variants correlates with their tendency to aggregate in solution. FEBS Letters 1997, 418, 297-300, 10.1016/s0014-5793(97)01398-7.
- Catarina S. H. Jesus; Zaida L. Almeida; Daniela C. Vaz; Tiago Q. Faria; Rui M. M. Brito; A New Folding Kinetic Mechanism for Human Transthyretin and the Influence of the Amyloidogenic V30M Mutation. International Journal of Molecular Sciences 2016, 17, 1428, 10.3390/ijms17091428.
- Kazufumi Takano; Jun Funahashi; Katsuhide Yutani; The stability and folding process of amyloidogenic mutant human lysozymes. JBIC Journal of Biological Inorganic Chemistry 2001, 268, 155-159, 10.1046/j.1432-1327.2001.01863.x.
- Rivka Leah Isaacson; Alan G. Weeds; Alan R. Fersht; Equilibria and kinetics of folding of gelsolin domain 2 and mutants involved in familial amyloidosis-Finnish type. Proceedings of the National Academy of Sciences 1999, 96, 11247-11252, 10.1073/pnas.96.20.11247.
- Marianne A. Grant; Noel Lazo; Aleksey Lomakin; Margaret M. Condron; Hiromi Arai; Ghiam Yamin; Alan C. Rigby; David B. Teplow; Familial Alzheimer's disease mutations alter the stability of the amyloid beta-protein monomer folding nucleus. Proceedings of the National Academy of Sciences 2007, 104, 16522-16527, 10.1073/pnas.0705197104.
- Christopher M. Dobson; Protein misfolding, evolution and disease. Trends in Biochemical Sciences 1999, 24, 329-332, 10.1016/s0968-0004(99)01445-0.
- Christopher M. Dobson; Protein folding and misfolding. Nature 2003, 426, 884-890, 10.1038/nature02261.
- Fabrizio Chiti; Christopher M. Dobson; Protein Misfolding, Functional Amyloid, and Human Disease. Annual Review of Biochemistry 2006, 75, 333-366, 10.1146/annurev.biochem.75.101304.123901.
- Douglas M. Fowler; Atanas V. Koulov; William E. Balch; Jeffery W. Kelly; Functional amyloid – from bacteria to humans. Trends in Biochemical Sciences 2007, 32, 217-224, 10.1016/j.tibs.2007.03.003.
- Chi L.L. Pham; Ann Kwan; Margaret Sunde; Functional amyloid: widespread in Nature, diverse in purpose. Essays in Biochemistry 2014, 56, 207-219, 10.1042/bse0560207.
- Daniel Otzen; Roland Riek; Functional Amyloids. Cold Spring Harbor Perspectives in Biology 2019, 11, a033860, 10.1101/cshperspect.a033860.
- Anamika Avni; Hema M. Swasthi; Anupa Majumdar; Samrat Mukhopadhyay; Intrinsically disordered proteins in the formation of functional amyloids from bacteria to humans. Progress in Molecular Biology and Translational Science 2019, 166, 109-143, 10.1016/bs.pmbts.2019.05.005.
- Jean D. Sipe; Alan S. Cohen; Review: History of the Amyloid Fibril. Journal of Structural Biology 2000, 130, 88-98, 10.1006/jsbi.2000.4221.
- O. Sumner Makin; Louise C. Serpell; Structures for amyloid fibrils. The FEBS Journal 2005, 272, 5950-5961, 10.1111/j.1742-4658.2005.05025.x.
- William Close; Matthias Neumann; Andreas Schmidt; Manuel Hora; Karthikeyan Annamalai; Matthias Schmidt; Bernd Reif; Volker Schmidt; Nikolaus Grigorieff; Marcus Fändrich; et al. Physical basis of amyloid fibril polymorphism. Nature Communications 2018, 9, 1-7, 10.1038/s41467-018-03164-5.
- Rebecca Nelson; Michael Sawaya; Melinda Balbirnie; Anders Østergaard Madsen; Christian Riekel; Robert Grothe; David Eisenberg; Structure of the cross-β spine of amyloid-like fibrils. Nature 2005, 435, 773-778, 10.1038/nature03680.
- Jalandoni-Buan, A.C.; Decena-Soliven, A.L.A.; Cao, E.P.; Barraquio, V.L.; Barraquio, W.L. Characterization and Identification of Congo Red Decolorizing Bacteria from Monocultures and Consortia. Philipp. J. Sci. 2010, 139, 71–78.
- Melanie R Nilsson; Techniques to study amyloid fibril formation in vitro. Methods 2004, 34, 151-160, 10.1016/j.ymeth.2004.03.012.
- William Klunk; Robert F. Jacob; R.Preston Mason; Quantifying Amyloid β-Peptide (Aβ) Aggregation Using the Congo Red-Aβ (CR–Aβ) Spectrophotometric Assay. Analytical Biochemistry 1999, 266, 66-76, 10.1006/abio.1998.2933.
- Robyn Eisert; Liseda Felau; Lesley R. Brown; Methods for enhancing the accuracy and reproducibility of Congo red and thioflavin T assays. Analytical Biochemistry 2006, 353, 144-146, 10.1016/j.ab.2006.03.015.
- Alexander J. Howie; Douglas B. Brewer; Optical properties of amyloid stained by Congo red: History and mechanisms. Micron 2008, 40, 285-301, 10.1016/j.micron.2008.10.002.
- Sait Sen; Gülçin Başdemir; Diagnosis of renal amyloidosis using Congo red fluorescence. Pathology International 2003, 53, 534-538, 10.1046/j.1440-1827.2003.01513.x.
- Tamar A. Giorgadze; Natsuko Shiina; Zubair W. Baloch; John E. Tomaszewski; Prabodh K. Gupta; Improved detection of amyloid in fat pad aspiration: An evaluation of Congo red stain by fluorescent microscopy. Diagnostic Cytopathology 2004, 31, 300-306, 10.1002/dc.20131.
- Harry Levine; Thioflavine T interaction with synthetic Alzheimer's diseaseβ-amyloid peptides: Detection of amyloid aggregation in solution. Protein Science 1993, 2, 404-410, 10.1002/pro.5560020312.
- Yair Porat; Adel Abramowitz; Ehud Gazit; Inhibition of Amyloid Fibril Formation by Polyphenols: Structural Similarity and Aromatic Interactions as a Common Inhibition Mechanism. Chemical Biology & Drug Design 2005, 67, 27-37, 10.1111/j.1747-0285.2005.00318.x.
- William G. Turnell; John T. Finch; Binding of the dye congo red to the amyloid protein pig insulin reveals a novel homology amongst amyloid-forming peptide sequences. Journal of Molecular Biology 1992, 227, 1205-1223, 10.1016/0022-2836(92)90532-o.
- Yong-Sung Kim; Theodore W. Randolph; Mark C. Manning; Fred J. Stevens; John F. Carpenter; Congo Red Populates Partially Unfolded States of an Amyloidogenic Protein to Enhance Aggregation and Amyloid Fibril Formation. Journal of Biological Chemistry 2003, 278, 10842-10850, 10.1074/jbc.m212540200.
- B Caughey; D Ernst; R E Race; Congo red inhibition of scrapie agent replication. Journal of Virology 1993, 67, 6270-6272, 10.1128/jvi.67.10.6270-6272.1993.
- A. Lorenzo; B. A. Yankner; Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo red.. Proceedings of the National Academy of Sciences 1994, 91, 12243-12247, 10.1073/pnas.91.25.12243.
- Harish Chander; Abha Chauhan; Ved Chauhan; Binding of Proteases to Fibrillar Amyloid-β Protein and its Inhibition by Congo Red. Journal of Alzheimer's Disease 2007, 12, 261-269, 10.3233/JAD-2007-12308.
- Vassar, P.S.; Culling, C.F. Fluorescent stains, with special reference to amyloid and connective tissues. Arch. Pathol. 1959, 68, 487–498.
- Kelényi, G. Thioflavin S fluorescent and Congo red anisotropic stainings in the histologic demonstration of amyloid. Acta Neuropathol. 1967, 7, 336–348.
- Nadine D. Younan; John H. Viles; A Comparison of Three Fluorophores for the Detection of Amyloid Fibers and Prefibrillar Oligomeric Assemblies. ThT (Thioflavin T); ANS (1-Anilinonaphthalene-8-sulfonic Acid); and bisANS (4,4′-Dianilino-1,1′-binaphthyl-5,5′-disulfonic Acid). Biochemistry 2015, 54, 4297-4306, 10.1021/acs.biochem.5b00309.
- Sureshbabu Nagarajan; Lisa J. Lapidus; Fluorescent Probe DCVJ Shows High Sensitivity for Characterization of Amyloid β-Peptide Early in the Lag Phase. ChemBioChem 2017, 18, 2205-2211, 10.1002/cbic.201700387.
- Rajesh Mishra; Daniel Sjölander; Per Hammarström; Spectroscopic characterization of diverse amyloid fibrils in vitro by the fluorescent dye Nile red. Mol. BioSyst. 2011, 7, 1232-1240, 10.1039/c0mb00236d.
- Vladyslava Kovalska; Svitlana Chernii; Mykhaylo Losytskyy; Iryna Tretyakova; Yan Dovbii; Alexandr Gorski; Victor Chernii; Rafal Czerwieniec; Sergiy Yarmoluk; Design of functionalized β-ketoenole derivatives as efficient fluorescent dyes for detection of amyloid fibrils. New Journal of Chemistry 2018, 42, 13308-13318, 10.1039/c8nj01020j.
- Adeline Marianne Fanni; Florencia A. Monge; Chia-Yu Lin; Arjun Thapa; Kiran Bhaskar; David G. Whitten; Eva Y. Chi; High Selectivity and Sensitivity of Oligomeric p-Phenylene Ethynylenes for Detecting Fibrillar and Prefibrillar Amyloid Protein Aggregates. ACS Chemical Neuroscience 2019, 10, 1813-1825, 10.1021/acschemneuro.8b00719.
- Sirvan Abbasbeigi; Hadi Adibi; Sajad Moradi; Seyyed Abolghasem Ghadami; Reza Khodarahmi; Detection/quantification of amyloid aggregation in solution using the novel fluorescent benzofuranone-derivative compounds as amyloid fluorescent probes: synthesis and in vitro characterization. Journal of the Iranian Chemical Society 2019, 16, 1225-1237, 10.1007/s13738-019-01599-1.
- Adam Crystal; Benoit I. Giasson; Alexander Crowe; Mei-Ping Kung; Zhi-Ping Zhuang; John Q. Trojanowski; Virginia M.-Y. Lee; A comparison of amyloid fibrillogenesis using the novel fluorescent compound K114. Journal of Neurochemistry 2003, 86, 1359-1368, 10.1046/j.1471-4159.2003.01949.x.
- Marie L. Schmidt; Theresa Schuck; Shelly Sheridan; Mei-Ping Kung; Hank Kung; Zhi-Ping Zhuang; Catherine Bergeron; Jacque S. Lamarche; Daniel Skovronsky; Benoit I. Giasson; et al.Virginia M.-Y. LeeJohn Q. Trojanowski The Fluorescent Congo Red Derivative, (Trans, Trans)−1-Bromo-2,5-Bis-(3-Hydroxycarbonyl-4-Hydroxy)Styrylbenzene (BSB), Labels Diverse β-Pleated Sheet Structures in Postmortem Human Neurodegenerative Disease Brains. The American Journal of Pathology 2001, 159, 937-943, 10.1016/s0002-9440(10)61769-5.
- Scot D. Styren; Ronald L. Hamilton; Gisele C. Styren; William E. Klunk; X-34, A Fluorescent Derivative of Congo Red: A Novel Histochemical Stain for Alzheimer's Disease Pathology. Journal of Histochemistry & Cytochemistry 2000, 48, 1223-1232, 10.1177/002215540004800906.
- Xiao-Peng He; Qiong Deng; Liang Cai; Chang-Zheng Wang; Yi Zang; Jia Li; Guo-Rong Chen; He Tian; Fluorogenic Resveratrol-Confined Graphene Oxide For Economic and Rapid Detection Of Alzheimer’s Disease. ACS Applied Materials & Interfaces 2014, 6, 5379-5382, 10.1021/am5010909.
- K.D. Volkova; V.B. Kovalska; A.O. Balanda; Mykhaylo Losytskyy; A.G. Golub; R.J. Vermeij; Vinod Subramaniam; O.I. Tolmachev; S.M. Yarmoluk; Specific fluorescent detection of fibrillar α-synuclein using mono- and trimethine cyanine dyes. Bioorganic & Medicinal Chemistry 2008, 16, 1452-1459, 10.1016/j.bmc.2007.10.051.
- K.D. Volkova; V.B. Kovalska; A.O. Balanda; R.J. Vermeij; V. Subramaniam; Yu. L. Slominskii; S.M. Yarmoluk; Cyanine dye–protein interactions: Looking for fluorescent probes for amyloid structures. Journal of Biochemical and Biophysical Methods 2007, 70, 727-733, 10.1016/j.jbbm.2007.03.008.
- José Luna-Muñoz; Janneth Peralta-Ramirez; Laura Chávez-Macías; Charles R. Harrington; Claude M. Wischik; Raúl Mena; Thiazin red as a neuropathological tool for the rapid diagnosis of Alzheimer’s disease in tissue imprints. Acta Neuropathologica 2008, 116, 507-515, 10.1007/s00401-008-0431-x.
- Anna I. Sulatskaya; Maksim I. Sulatsky; Iuliia A. Antifeeva; Irina M. Kuznetsova; Konstantin K. Turoverov; Structural Analogue of Thioflavin T, DMASEBT, as a Tool for Amyloid Fibrils Study. Analytical Chemistry 2019, 91, 3131-3140, 10.1021/acs.analchem.8b05737.
- Matthew Biancalana; Shohei Koide; Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2010, 1804, 1405-1412, 10.1016/j.bbapap.2010.04.001.
- Yakupova, E.I.; Bobyleva, L.G.; Vikhlyantsev, I.M.; Bobylev, A.G. Congo Red and amyloids: History and relationship. Biosci. Rep. 2019, 39.
- Merrill D. Benson; Joel N. Buxbaum; David S. Eisenberg; Giampaolo Merlini; Maria J. M. Saraiva; Yoshiki Sekijima; Jean D. Sipe; Per Westermark; Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid 2018, 25, 215-219, 10.1080/13506129.2018.1549825.
- A.J. Geddes; K.D. Parker; E.D.T. Atkins; E. Beighton; “Cross-β” conformation in proteins. Journal of Molecular Biology 1968, 32, 343-358, 10.1016/0022-2836(68)90014-4.
- M Sunde; C Blake; The Structure of Amyloid Fibrils by Electron Microscopy and X-Ray Diffraction. Advances in Protein Chemistry Volume 12 1997, 50, 123-159, 10.1016/s0065-3233(08)60320-4.
- Ravindra Kodali; Ronald Wetzel; Polymorphism in the intermediates and products of amyloid assembly. Current Opinion in Structural Biology 2007, 17, 48-57, 10.1016/j.sbi.2007.01.007.
- E. D. Eanes; G. G. Glenner; X-RAY DIFFRACTION STUDIES ON AMYLOID FILAMENTS. Journal of Histochemistry & Cytochemistry 1968, 16, 673-677, 10.1177/16.11.673.
- L. Bonar; A. S. Cohen; M. M. Skinner; Characterization of the Amyloid Fibril as a Cross- Protein. Experimental Biology and Medicine 1969, 131, 1373-1375, 10.3181/00379727-131-34110.
- Margaret Sunde; Louise Serpell; Mark Bartlam; Paul E Fraser; Mark B Pepys; Colin C.F Blake; Common core structure of amyloid fibrils by synchrotron X-ray diffraction. Journal of Molecular Biology 1997, 273, 729-739, 10.1006/jmbi.1997.1348.
- Rebecca Nelson; David Eisenberg; Recent atomic models of amyloid fibril structure. Current Opinion in Structural Biology 2006, 16, 260-265, 10.1016/j.sbi.2006.03.007.
- M. I. Ivanova; M. J. Thompson; D. Eisenberg; A systematic screen of beta2-microglobulin and insulin for amyloid-like segments. Proceedings of the National Academy of Sciences 2006, 103, 4079-4082, 10.1073/pnas.0511298103.
- David S. Eisenberg; Michael R. Sawaya; Structural Studies of Amyloid Proteins at the Molecular Level. Annual Review of Biochemistry 2017, 86, 69-95, 10.1146/annurev-biochem-061516-045104.
- Shilpa Sambashivan; Yanshun Liu; Michael Sawaya; Mari Gingery; David Eisenberg; Amyloid-like fibrils of ribonuclease A with three-dimensional domain-swapped and native-like structure. Nature 2005, 437, 266-269, 10.1038/nature03916.
- Marcus Fändrich; Jessica Meinhardt; Nikolaus Grigorieff; Structural polymorphism of Alzheimer Aβ and other amyloid fibrils. Prion 2009, 3, 89-93, 10.4161/pri.3.2.8859.
- Jessica Meinhardt; Carsten Sachse; Peter Hortschansky; Nikolaus Grigorieff; Marcus Fändrich; Aβ(1-40) Fibril Polymorphism Implies Diverse Interaction Patterns in Amyloid Fibrils. Journal of Molecular Biology 2009, 386, 869-877, 10.1016/j.jmb.2008.11.005.
- Karolina L. Zapadka; Frederik J. Becher; A. L. Gomes Dos Santos; Sophie E. Jackson; Factors affecting the physical stability (aggregation) of peptide therapeutics. Interface Focus 2017, 7, 20170030-20170030, 10.1098/rsfs.2017.0030.
- Oscar Conchillo-Solé; Natalia S de Groot; Francesc X Avilés; Josep Vendrell; Xavier Daura; Salvador Ventura; AGGRESCAN: a server for the prediction and evaluation of "hot spots" of aggregation in polypeptides. BMC Bioinformatics 2007, 8, 65-65, 10.1186/1471-2105-8-65.
- Natalia S. de Groot; Virginia Castillo; Ricardo Graña-Montes; Salvador Ventura; AGGRESCAN: Method, Application, and Perspectives for Drug Design. Methods in Molecular Biology 2011, 819, 199-220, 10.1007/978-1-61779-465-0_14.
- Rafael Zambrano; Michal Jamroz; Agata Szczasiuk; Jordi Pujols; Sebastian Kmiecik; Salvador Ventura; AGGRESCAN3D (A3D): server for prediction of aggregation properties of protein structures. Nucleic Acids Research 2015, 43, W306-W313, 10.1093/nar/gkv359.
- Jordi Pujols; Samuel Peña-Díaz; Salvador Ventura; AGGRESCAN3D: Toward the Prediction of the Aggregation Propensities of Protein Structures. Mitochondrial Bioenergetics 2018, 1762, 427-443, 10.1007/978-1-4939-7756-7_21.
- Pawel P. Wozniak; Malgorzata Kotulska; AmyLoad: website dedicated to amyloidogenic protein fragments. Bioinformatics 2015, 31, 3395-3397, 10.1093/bioinformatics/btv375.
- Burdukiewicz, M.; Sobczyk, P.; Rödiger, S.; Duda-Madej, A.; Mackiewicz, P.; Kotulska, M. Amyloidogenic motifs revealed by n-gram analysis. Sci. Rep. 2017, 7, 12961.
- Charles W. O'Donnell; Jérôme Waldispühl; Mieszko Lis; Randal Halfmann; Srinivas Devadas; Susan Lindquist; Bonnie Berger; A method for probing the mutational landscape of amyloid structure. Bioinformatics 2011, 27, i34-i42, 10.1093/bioinformatics/btr238.
- Kimon K Frousios; Vassiliki A Iconomidou; Carolina-Maria Karletidi; Stavros J Hamodrakas; Amyloidogenic determinants are usually not buried. BMC Structural Biology 2009, 9, 44-44, 10.1186/1472-6807-9-44.
- Antonios Tsolis; Nikos Papandreou; Vassiliki A. Iconomidou; Stavros J. Hamodrakas; A Consensus Method for the Prediction of ‘Aggregation-Prone’ Peptides in Globular Proteins. PLOS ONE 2013, 8, e54175, 10.1371/journal.pone.0054175.
- Carlos Familia; Sarah R. Dennison; Alexandre Quintas; David A. Phoenix; Prediction of Peptide and Protein Propensity for Amyloid Formation. PLoS ONE 2015, 10, e0134679, 10.1371/journal.pone.0134679.
- Allen W. Bryan; Matthew Menke; Lenore J. Cowen; Susan L. Lindquist; Bonnie Berger; BETASCAN: Probable β-amyloids Identified by Pairwise Probabilistic Analysis. PLoS Computational Biology 2009, 5, e1000333, 10.1371/journal.pcbi.1000333.
- Melissa J. Landrum; Jennifer M. Lee; Mark Benson; Garth Brown; Chen Chao; Shanmuga Chitipiralla; Baoshan Gu; Jennifer Hart; Uglas Hoffman; Jeffrey Hoover; et al.Wonhee JangKenneth KatzMichael OvetskyGeorge RileyAmanjeev SethiRay TullyRicardo Villamarin-SalomonWendy RubinsteinNna R. Maglott ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Research 2015, 44, D862-D868, 10.1093/nar/gkv1222 [doi].
- Hamed Tabatabaei Ghomi; Elizabeth M. Topp; Markus A. Lill; Fibpredictor: a computational method for rapid prediction of amyloid fibril structures. Journal of Molecular Modeling 2016, 22, 206, 10.1007/s00894-016-3066-1.
- Pawel Gasior; Malgorzata Kotulska; FISH Amyloid – a new method for finding amyloidogenic segments in proteins based on site specific co-occurence of aminoacids. BMC Bioinformatics 2014, 15, 54-54, 10.1186/1471-2105-15-54.
- Sergiy Garbuzynskiy; Michail Yu. Lobanov; Oxana V. Galzitskaya; FoldAmyloid: a method of prediction of amyloidogenic regions from protein sequence. Bioinformatics 2009, 26, 326-332, 10.1093/bioinformatics/btp691.
- A. Mary Thangakani; Sandeep Kumar; R. Nagarajan; D. Velmurugan; M. Michael Gromiha; GAP: towards almost 100 percent prediction for β-strand-mediated aggregating peptides with distinct morphologies. Bioinformatics 2014, 30, 1983-1990, 10.1093/bioinformatics/btu167.
- Taner Z. Sen; Robert L. Jernigan; Jean Garnier; Andrzej Kloczkowski; GOR V server for protein secondary structure prediction. Bioinformatics 2005, 21, 2787-2788, 10.1093/bioinformatics/bti408.
- Maksim Kouza; Eshel Faraggi; Andrzej Kolinski; Andrzej Kloczkowski; The GOR Method of Protein Secondary Structure Prediction and Its Application as a Protein Aggregation Prediction Tool. Springer Protocols Handbooks 2016, 1484, 7-24, 10.1007/978-1-4939-6406-2_2.
- Mathieu Emily; Anthony Talvas; Christian Delamarche; MetAmyl: A METa-Predictor for AMYLoid Proteins. PLoS ONE 2013, 8, e79722, 10.1371/journal.pone.0079722.
- Farzeen Munir; Sadaf Gull; Amina Asif; Fayyaz-Ul-Amir Afsar Minhas; MILAMP: Multiple Instance Prediction of Amyloid Proteins. IEEE/ACM Transactions on Computational Biology and Bioinformatics 2019, 18, 1142-1150, 10.1109/tcbb.2019.2936846.
- Changsik Kim; Jiwon Choi; Seong Joon Lee; William J. Welsh; Sukjoon Yoon; NetCSSP: web application for predicting chameleon sequences and amyloid fibril formation. Nucleic Acids Research 2009, 37, W469-W473, 10.1093/nar/gkp351.
- Antonio Trovato; Fabrizio Chiti; Amos Maritan; Flavio Seno; Insight into the Structure of Amyloid Fibrils from the Analysis of Globular Proteins. PLOS Computational Biology 2006, 2, e170, 10.1371/journal.pcbi.0020170.
- Antonio Trovato; Flavio Seno; Silvio C.E. Tosatto; The PASTA server for protein aggregation prediction. Protein Engineering Design and Selection 2007, 20, 521-523, 10.1093/protein/gzm042.
- Ian Walsh; Flavio Seno; Silvio C.E. Tosatto; Antonio Trovato; PASTA 2.0: an improved server for protein aggregation prediction. Nucleic Acids Research 2014, 42, W301-W307, 10.1093/nar/gku399.
- Mengting Niu; Yanjuan Li; Chunyu Wang; Ke Han; RFAmyloid: A Web Server for Predicting Amyloid Proteins. International Journal of Molecular Sciences 2018, 19, 2071, 10.3390/ijms19072071.
- G. De Baets; J. Van Durme; Joke Reumers; Sebastian Maurer-Stroh; P. Vanhee; J. Dopazo; J. Schymkowitz; F. Rousseau; SNPeffect 4.0: on-line prediction of molecular and structural effects of protein-coding variants. Nucleic Acids Research 2011, 40, D935-D939, 10.1093/nar/gkr996.
- Allen W. Bryan Jr.; Colm O'Donnell; Matthew Menke; Lenore J. Cowen; Susan Lindquist; Bonnie Berger; STITCHER: Dynamic assembly of likely amyloid and prion β‐structures from secondary structure predictions. Proteins: Structure, Function, and Bioinformatics 2011, 80, 410-420, 10.1002/prot.23203.
- Ana-Maria Fernandez-Escamilla; Frederic Rousseau; Joost Schymkowitz; Luis Serrano; Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nature Biotechnology 2004, 22, 1302-1306, 10.1038/nbt1012 nbt1012 [pii].
- Sebastian Maurer-Stroh; Maja Debulpaep; Nico Kuemmerer; Manuela Lopez De La Paz; Ivo Martins; Joke Reumers; Kyle L Morris; Alastair Copland; Louise Serpell; Luis Serrano; et al.Joost SchymkowitzFrederic Rousseau Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nature Methods 2010, 7, 237-242, 10.1038/nmeth.1432.
- Jacinte Beerten; Joost Van Durme; Rodrigo Gallardo; Emidio Capriotti; Louise Serpell; Frederic Rousseau; Joost Schymkowitz; WALTZ-DB: a benchmark database of amyloidogenic hexapeptides. Bioinformatics 2015, 31, 1698-1700, 10.1093/bioinformatics/btv027.
- Nikolaos Louros; Katerina Konstantoulea; Matthias De Vleeschouwer; Meine Ramakers; Joost Schymkowitz; Frederic Rousseau; WALTZ-DB 2.0: an updated database containing structural information of experimentally determined amyloid-forming peptides. Nucleic Acids Research 2019, 48, D389-D393, 10.1093/nar/gkz758.
- Michael J. Thompson; Stuart A. Sievers; John Karanicolas; Magdalena I. Ivanova; David Baker; David Eisenberg; The 3D profile method for identifying fibril-forming segments of proteins. Proceedings of the National Academy of Sciences 2006, 103, 4074-4078, 10.1073/pnas.0511295103.
- Gian Gaetano Tartaglia; Michele Vendruscolo; The Zyggregator method for predicting protein aggregation propensities. Chemical Society Reviews 2008, 37, 1395-401, 10.1039/b706784b.
- Michael GroB; Proteins that Convert from a Helix to b Sheet Implications for Folding and Disease. Current Protein & Peptide Science 2000, 1, 339-347, 10.2174/1389203003381289.
- Charlotte Hauser; R. Deng; A. Mishra; Y. Loo; U. Khoe; F. Zhuang; D. W. Cheong; A. Accardo; Michael Sullivan; C. Riekel; et al.Jackie YingU. A. Hauser Natural tri- to hexapeptides self-assemble in water to amyloid -type fiber aggregates by unexpected -helical intermediate structures. Proceedings of the National Academy of Sciences 2011, 108, 1361-1366, 10.1073/pnas.1014796108.
- Dhiman Ghosh; Pradeep K. Singh; Shruti Sahay; Narendra Nath Jha; Reeba Jacob; Shamik Sen; Ashutosh Kumar; Roland Riek; Samir K. Maji; Structure based aggregation studies reveal the presence of helix-rich intermediate during α-Synuclein aggregation. Scientific Reports 2015, 5, 9228-9228, 10.1038/srep09228.
- Katarzyna Cieślik-Boczula; Alpha-helix to beta-sheet transition in long-chain poly- l -lysine: Formation of alpha-helical fibrils by poly- l -lysine. Biochimie 2017, 137, 106-114, 10.1016/j.biochi.2017.03.006.
- Ming Ni; Shuangmu Zhuo; Ciprian Iliescu; Peter T. C. So; Jodhbir S. Mehta; Hanry Yu; Charlotte A. E. Hauser; Self‐assembling amyloid‐like peptides as exogenous second harmonic probes for bioimaging applications. Journal of Biophotonics 2019, 12, e201900065, 10.1002/jbio.201900065.
- Einav Tayeb-Fligelman; Orly Tabachnikov; Asher Moshe; Orit Goldshmidt-Tran; Michael R. Sawaya; Nicolas Coquelle; Jacques-Philippe Colletier; Meytal Landau; The cytotoxic Staphylococcus aureus PSMα3 reveals a cross-α amyloid-like fibril. Science 2017, 355, 831-833, 10.1126/science.aaf4901.
- Luc Bousset; Neil Thomson; Sheena E. Radford; Ronald Melki; The yeast prion Ure2p retains its native α-helical conformation upon assembly into protein fibrils in vitro. The EMBO Journal 2002, 21, 2903-2911, 10.1093/emboj/cdf303.
- K. S. Taylor; M.-Z. Lou; T.-M. Chin; N. C. Yang; R. M. Garavito; A novel, multilayer structure of a helical peptide. Protein Science 1996, 5, 414-421, 10.1002/pro.5560050302.
- Gilbert G. Privé; Daniel H. Anderson; Laura Wesson; Duilio Cascio; David Eisenberg; Packed protein bilayers in the 0.90 å resolution structure of a designed alpha helical bundle. Protein Science 1999, 8, 1400-1409, 10.1110/ps.8.7.1400.
- Sudipta Mondal; Lihi Adler-Abramovich; Ayala Lampel; Yaron Bram; Sophia Lipstman; Ehud Gazit; Formation of functional super-helical assemblies by constrained single heptad repeat. Nature Communications 2015, 6, 8615, 10.1038/ncomms9615.
- Tj Brunette; Fabio Parmeggiani; Po-Ssu Huang; Gira Bhabha; Damian C. Ekiert; Susan E. Tsutakawa; Greg L. Hura; John A. Tainer; David Baker; Exploring the repeat protein universe through computational protein design. Nature 2015, 528, 580-584, 10.1038/nature16162.
- Pham, C.L.; Shanmugam, N.; Strange, M.; O’Carroll, A.; Brown, J.W.; Sierecki, E.; Gambin, Y.; Steain, M.; Sunde, M. Viral M45 and necroptosis-associated proteins form heteromeric amyloid assemblies. EMBO Rep. 2019, 20.
- Miguel Mompeán; Wenbo Li; Jixi Li; Ségolène Laage; Ansgar B. Siemer; Gunes Bozkurt; Hao Wu; Ann E. McDermott; The Structure of the Necrosome RIPK1-RIPK3 Core, a Human Hetero-Amyloid Signaling Complex. Cell 2018, 173, 1244-1253.e10, 10.1016/j.cell.2018.03.032.
- Brian O'Nuallain; Angela D. Williams; Per Westermark; Ronald Wetzel; Seeding Specificity in Amyloid Growth Induced by Heterologous Fibrils. Journal of Biological Chemistry 2004, 279, 17490-17499, 10.1074/jbc.m311300200.
- Marie E. Oskarsson; Johan F Paulsson; Sebastian W. Schultz; Martin Ingelsson; Per Westermark; Gunilla T. Westermark; In Vivo Seeding and Cross-Seeding of Localized Amyloidosis. The American Journal of Pathology 2015, 185, 834-846, 10.1016/j.ajpath.2014.11.016.
- Rodrigo Morales; Lisbell D. Estrada; Rodrigo Diaz-Espinoza; Diego Morales-Scheihing; Maria C. Jara; Joaquin Castilla; Claudio Soto; Molecular Cross Talk between Misfolded Proteins in Animal Models of Alzheimer's and Prion Diseases. The Journal of Neuroscience 2010, 30, 4528-4535, 10.1523/JNEUROSCI.5924-09.2010.
- Janett Köppen; Anja Schulze; Lisa Machner; Michael Wermann; Rico Eichentopf; Max Guthardt; Angelika Hähnel; Jessica Klehm; Marie-Christin Kriegeskorte; Maike Hartlage-Rübsamen; et al.Markus MorawskiStephan Von HörstenHans-Ulrich DeMuthSteffen RoßnerStephan Schilling Amyloid-Beta Peptides Trigger Aggregation of Alpha-Synuclein In Vitro. Molecules 2020, 25, 580, 10.3390/molecules25030580.
- Gotz, J.; Chen, F.; van Dorpe, J.; Nitsch, R.M. Formation of Neurofibrillary Tangles in P301L Tau Transgenic Mice Induced by Abeta 42 Fibrils. Science 2001, 293, 1491–1495.
- Cristian A. Lasagna-Reeves; Diana L. Castillo-Carranza; Marcos J. Guerrero-Muñoz; George R. Jackson; Rakez Kayed; Preparation and Characterization of Neurotoxic Tau Oligomers. Biochemistry 2010, 49, 10039-10041, 10.1021/bi1016233.
- Karishma Bhasne; Sanjana Sebastian; Neha Jain; Samrat Mukhopadhyay; Synergistic Amyloid Switch Triggered by Early Heterotypic Oligomerization of Intrinsically Disordered α-Synuclein and Tau. Journal of Molecular Biology 2018, 430, 2508-2520, 10.1016/j.jmb.2018.04.020.
- K. Lundmark; G. T. Westermark; A. Olsen; P. Westermark; Protein fibrils in nature can enhance amyloid protein A amyloidosis in mice: Cross-seeding as a disease mechanism. Proceedings of the National Academy of Sciences 2005, 102, 6098-6102, 10.1073/pnas.0501814102.
- I. L. Derkatch; S. M. Uptain; T. F. Outeiro; Rajaraman Krishnan; S. L. Lindquist; S. W. Liebman; Effects of Q/N-rich, polyQ, and non-polyQ amyloids on the de novo formation of the [PSI+] prion in yeast and aggregation of Sup35 in vitro. Proceedings of the National Academy of Sciences 2004, 101, 12934-12939, 10.1073/pnas.0404968101.
- Neal Hammer; J. C. Schmidt; M. R. Chapman; The curli nucleator protein, CsgB, contains an amyloidogenic domain that directs CsgA polymerization. Proceedings of the National Academy of Sciences 2007, 104, 12494-12499, 10.1073/pnas.0703310104.
- Kyle L. Morris; Louise C. Serpell; X-Ray Fibre Diffraction Studies of Amyloid Fibrils. Springer Protocols Handbooks 2012, 849, 121-135, 10.1007/978-1-61779-551-0_9.
- Robert Tycko; Solid-State NMR Studies of Amyloid Fibril Structure. Annual Review of Physical Chemistry 2011, 62, 279-299, 10.1146/annurev-physchem-032210-103539.
- Louise C Serpell; Judith M Smith; Direct visualisation of the β-sheet structure of synthetic Alzheimer’s amyloid. Journal of Molecular Biology 2000, 299, 225-231, 10.1006/jmbi.2000.3650.
- Rachel W. Martin; John E. Kelly; Jessica I. Kelz; Advances in instrumentation and methodology for solid-state NMR of biological assemblies. Journal of Structural Biology 2018, 206, 73-89, 10.1016/j.jsb.2018.09.003.
- Beat H. Meier; Roland Riek; Anja Böckmann; Emerging Structural Understanding of Amyloid Fibrils by Solid-State NMR. Trends in Biochemical Sciences 2017, 42, 777-787, 10.1016/j.tibs.2017.08.001.
- Xiao-Chen Bai; Greg McMullan; Sjors H.W Scheres; How cryo-EM is revolutionizing structural biology. Trends in Biochemical Sciences 2015, 40, 49-57, 10.1016/j.tibs.2014.10.005.
- Alan S. Cohen; Evan Calkins; Electron Microscopic Observations on a Fibrous Component in Amyloid of Diverse Origins. Nature 1959, 183, 1202-1203, 10.1038/1831202a0.
- Werner Kühlbrandt; The Resolution Revolution. Science 2014, 343, 1443-1444, 10.1126/science.1251652.
- Lilia Milanesi; Tania Sheynis; Wei-Feng Xue; Elena Orlova; Andrew Hellewell; Raz Jelinek; Eric Hewitt; Sheena Radford; Helen R. Saibil; Direct three-dimensional visualization of membrane disruption by amyloid fibrils. Proceedings of the National Academy of Sciences 2012, 109, 20455-20460, 10.1073/pnas.1206325109.
- Kevin W. Tipping; Patricija van Oosten-Hawle; Eric Hewitt; Sheena E. Radford; Amyloid Fibres: Inert End-Stage Aggregates or Key Players in Disease?. Trends in Biochemical Sciences 2015, 40, 719-727, 10.1016/j.tibs.2015.10.002.
- Vibha Taneja; Meenakshi Verma; Abhishek Vats; Toxic species in amyloid disorders: Oligomers or mature fibrils. Annals of Indian Academy of Neurology 2015, 18, 138-45, 10.4103/0972-2327.144284.
- Elisa Evangelisti; Roberta Cascella; Matteo Becatti; Giovanna Marrazza; Christopher M. Dobson; Fabrizio Chiti; Massimo Stefani; Cristina Cecchi; Binding affinity of amyloid oligomers to cellular membranes is a generic indicator of cellular dysfunction in protein misfolding diseases. Scientific Reports 2016, 6, 32721, 10.1038/srep32721.
- Mirella Vivoli Vega; Roberta Cascella; Serene W Chen; Giuliana Fusco; Alfonso De Simone; Christopher M. Dobson; Cristina Cecchi; Fabrizio Chiti; The Toxicity of Misfolded Protein Oligomers Is Independent of Their Secondary Structure. ACS Chemical Biology 2019, 14, 1593-1600, 10.1021/acschembio.9b00324.
- Heidi Olzscha; Sonya M. Schermann; Andreas C. Woerner; Stefan Pinkert; Michael H. Hecht; Gian G. Tartaglia; Michele Vendruscolo; Manajit Hayer-Hartl; F. Ulrich Hartl; R. Martin Vabulas; et al. Amyloid-like Aggregates Sequester Numerous Metastable Proteins with Essential Cellular Functions. Cell 2011, 144, 67-78, 10.1016/j.cell.2010.11.050.
- Benedetta Mannini; Estefania Mulvihill; Caterina Sgromo; Roberta Cascella; Reza Khodarahmi; Matteo Ramazzotti; Christopher M. Dobson; Cristina Cecchi; Fabrizio Chiti; Toxicity of Protein Oligomers Is Rationalized by a Function Combining Size and Surface Hydrophobicity. ACS Chemical Biology 2014, 9, 2309-2317, 10.1021/cb500505m.
- Benedetta Mannini; R. Cascella; M. Zampagni; M. van Waarde-Verhagen; S. Meehan; Cintia Roodveldt; S. Campioni; M. Boninsegna; A. Penco; A. Relini; et al.Harm KampingaC. M. DobsonMark WilsonCristina CecchiF. Chiti Molecular mechanisms used by chaperones to reduce the toxicity of aberrant protein oligomers. Proceedings of the National Academy of Sciences 2012, 109, 12479-12484, 10.1073/pnas.1117799109.
- Monica Bucciantini; Elisa Giannoni; Fabrizio Chiti; Fabiana Baroni; Lucia Formigli; Jesús Zurdo; Niccolò Taddei; Giampietro Ramponi; Christopher M. Dobson; Massimo Stefani; et al. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507-511, 10.1038/416507a.
- Joanne McLaurin; Avi Chakrabartty; Membrane Disruption by Alzheimer β-Amyloid Peptides Mediated through Specific Binding to Either Phospholipids or Gangliosides. Journal of Biological Chemistry 1996, 271, 26482-26489, 10.1074/jbc.271.43.26482.
- Iryna Benilova; Eric Karran; Bart De Strooper; The toxic Aβ oligomer and Alzheimer's disease: an emperor in need of clothes. Nature Neuroscience 2012, 15, 349-357, 10.1038/nn.3028.
- Sophia C. Goodchild; Tania Sheynis; Rebecca Thompson; Kevin W. Tipping; Wei-Feng Xue; Neil A. Ranson; Paul A. Beales; Eric W. Hewitt; Sheena E. Radford; β2-Microglobulin Amyloid Fibril-Induced Membrane Disruption Is Enhanced by Endosomal Lipids and Acidic pH. PLOS ONE 2014, 9, e104492, 10.1371/journal.pone.0104492.
- Konstanze F. Winklhofer; Christian Haass; Mitochondrial dysfunction in Parkinson's disease. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2010, 1802, 29-44, 10.1016/j.bbadis.2009.08.013.
- Hazel L. Roberts; David R. Brown; Seeking a Mechanism for the Toxicity of Oligomeric α-Synuclein. Biomolecules 2015, 5, 282-305, 10.3390/biom5020282.
- Thomas C.T. Michaels; Anđela Šarić; Johnny Habchi; Sean Chia; Georg Meisl; Michele Vendruscolo; Christopher M. Dobson; Tuomas P.J. Knowles; Chemical Kinetics for Bridging Molecular Mechanisms and Macroscopic Measurements of Amyloid Fibril Formation. Annual Review of Physical Chemistry 2018, 69, 273-298, 10.1146/annurev-physchem-050317-021322.
- Carl Frieden; Protein aggregation processes: In search of the mechanism. Protein Science 2007, 16, 2334-2344, 10.1110/ps.073164107.
- Alexander K. Buell; Christopher M. Dobson; Tuomas P.J. Knowles; The physical chemistry of the amyloid phenomenon: thermodynamics and kinetics of filamentous protein aggregation. Essays in Biochemistry 2014, 56, 11-39, 10.1042/bse0560011.
- Joseph T. Jarrett; Jr. Peter T. Lansbury; Amyloid fibril formation requires a chemically discriminating nucleation event: studies of an amyloidogenic sequence from the bacterial protein OsmB. Biochemistry 1992, 31, 12345-12352, 10.1021/bi00164a008.
- Walraj S. Gosal; Isobel J. Morten; Eric W. Hewitt; D. Alastair Smith; Neil H. Thomson; Sheena E. Radford; Competing Pathways Determine Fibril Morphology in the Self-assembly of β2-Microglobulin into Amyloid. Journal of Molecular Biology 2005, 351, 850-864, 10.1016/j.jmb.2005.06.040.
- Gemma Soldi; Francesco Bemporad; Silvia Torrassa; Annalisa Relini; Matteo Ramazzotti; Niccolò Taddei; Fabrizio Chiti; Amyloid Formation of a Protein in the Absence of Initial Unfolding and Destabilization of the Native State. Biophysical Journal 2005, 89, 4234-4244, 10.1529/biophysj.105.067538.
- Georgia Plakoutsi; Francesco Bemporad; Martino Calamai; Niccolò Taddei; Christopher M. Dobson; Fabrizio Chiti; Evidence for a Mechanism of Amyloid Formation Involving Molecular Reorganisation within Native-like Precursor Aggregates. Journal of Molecular Biology 2005, 351, 910-922, 10.1016/j.jmb.2005.06.043.
- Francesco Bemporad; Tommaso Vannocci; Lorena Varela; Ana I. Azuaga; Fabrizio Chiti; A model for the aggregation of the acylphosphatase from Sulfolobus solfataricus in its native-like state. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2008, 1784, 1986-1996, 10.1016/j.bbapap.2008.08.021.
- Javier Garcia-Pardo; Ricardo Graña-Montes; Marc Fernandez-Mendez; Angels Ruyra; Nerea Roher; Francesc Xavier Aviles; Julia Lorenzo; Salvador Ventura; Amyloid Formation by Human Carboxypeptidase D Transthyretin-like Domain under Physiological Conditions. Journal of Biological Chemistry 2014, 289, 33783-33796, 10.1074/jbc.m114.594804.
- Stephen W. Raso; Jeff Abel; Jesse M. Barnes; Kevin M. Maloney; Gary Pipes; Michael J. Treuheit; Jonathan King; David N. Brems; Aggregation of granulocyte-colony stimulating factor in vitro involves a conformationally altered monomeric state. Protein Science 2005, 14, 2246-2257, 10.1110/ps.051489405.
- Brent S. Kendrick; John F. Carpenter; Jeffrey L. Cleland; Theodore W. Randolph; A transient expansion of the native state precedes aggregation of recombinant human interferon. Proceedings of the National Academy of Sciences 1998, 95, 14142-14146, 10.1073/pnas.95.24.14142.
- Alexandre Quintas; Maria João Saraiva; Rui M. M. Brito; The Tetrameric Protein Transthyretin Dissociates to a Non-native Monomer in Solution. Journal of Biological Chemistry 1999, 274, 32943-32949, 10.1074/jbc.274.46.32943.
- Alexandre Quintas; Daniela Barroso De Moura Cipreste Vaz; Isabel Cardoso; Maria João Saraiva; Rui Brito; Tetramer Dissociation and Monomer Partial Unfolding Precedes Protofibril Formation in Amyloidogenic Transthyretin Variants. Journal of Biological Chemistry 2001, 276, 27207-27213, 10.1074/jbc.m101024200.
- David R. Booth; Margaret Sunde; Vittorio Bellotti; Carol V. Robinson; Winston L. Hutchinson; Paul E. Fraser; Philip N. Hawkins; Christopher M. Dobson; Sheena Radford; Colin C. F. Blake; et al.Mark B. Pepys Instability, unfolding and aggregation of human lysozyme variants underlying amyloid fibrillogenesis. Nature 1997, 385, 787-793, 10.1038/385787a0.
- J. Iñaki Guijarro; Margaret Sunde; Jonathan Jones; Iain D. Campbell; Christopher M. Dobson; Amyloid fibril formation by an SH3 domain. Proceedings of the National Academy of Sciences 1998, 95, 4224-4228, 10.1073/pnas.95.8.4224.
- Truscott, R.J.W.; Augusteyn, R.C. Oxidative changes in human lens proteins during senile nuclear cataract formation. Biochim. Et Biophys. Acta (BBA)-Protein Struct. 1977, 492, 43–52.
- Purnananda Guptasarma; Dorairajan Balasubramanian; Seichii Matsugo; Isao Saito; Hydroxyl radical mediated damage to proteins, with special reference to the crystallins. Biochemistry 1992, 31, 4296-4303, 10.1021/bi00132a021.
- Suzanne Hermeling; Huub Schellekens; Coen Maas; Martijn F.B.G. Gebbink; Daan J.A. Crommelin; Wim Jiskoot; Antibody Response to Aggregated Human Interferon Alpha2b in Wild-type and Transgenic Immune Tolerant Mice Depends on Type and Level of Aggregation. Journal of Pharmaceutical Sciences 2006, 95, 1084-1096, 10.1002/jps.20599.
- Hamid Mirzaei; Fred Regnier; Protein:protein aggregation induced by protein oxidation. Journal of Chromatography B 2008, 873, 8-14, 10.1016/j.jchromb.2008.04.025.
- Christian Landles; Kirupa Sathasivam; Andreas Weiss; Ben Woodman; Hilary Moffitt; Steve Finkbeiner; Banghua Sun; Juliette Gafni; Lisa M. Ellerby; Yvon Trottier; et al.William G. RichardsAlex OsmandPaolo PaganettiGillian P. Bates Proteolysis of Mutant Huntingtin Produces an Exon 1 Fragment That Accumulates as an Aggregated Protein in Neuronal Nuclei in Huntington Disease. Journal of Biological Chemistry 2010, 285, 8808-8823, 10.1074/jbc.m109.075028.
- Gary K.L. Chan; Andrzej Witkowski; Donald L. Gantz; Tianqi O. Zhang; Martin T. Zanni; Shobini Jayaraman; Giorgio Cavigiolio; Myeloperoxidase-mediated Methionine Oxidation Promotes an Amyloidogenic Outcome for Apolipoprotein A-I. Journal of Biological Chemistry 2015, 290, 10958-10971, 10.1074/jbc.m114.630442.
- M. A. Rosenfeld; V. B. Leonova; M. L. Konstantinova; S. D. Razumovskii; Self-assembly of fibrin monomers and fibrinogen aggregation during ozone oxidation. Biochemistry (Moscow) 2009, 74, 41-46, 10.1134/s0006297909010064.
- Alireza Roostaee; Sébastien Côté; Xavier Roucou; Aggregation and Amyloid Fibril Formation Induced by Chemical Dimerization of Recombinant Prion Protein in Physiological-like Conditions. Journal of Biological Chemistry 2009, 284, 30907-30916, 10.1074/jbc.m109.057950.
- Takumi Takata; Julie T. Oxford; Borries Demeler; Kirsten J. Lampi; Deamidation destabilizes and triggers aggregation of a lens protein, βA3-crystallin. Protein Science 2008, 17, 1565-1575, 10.1110/ps.035410.108.
- Yan Wei; Lan Chen; Ji Chen; Lin Ge; Rong Qiao He; Rapid glycation with D-ribose induces globular amyloid-like aggregations of BSA with high cytotoxicity to SH-SY5Y cells. BMC Cell Biology 2009, 10, 10-10, 10.1186/1471-2121-10-10.
- Lars Redecke; Stephan Binder; Mohammed I.Y. Elmallah; Rebecca Broadbent; Claudia Tilkorn; Benjamin Schulz; Patrick May; Arne Goos; Andreas Eich; Michael Rübhausen; et al.Christian Betzel UV-light-induced conversion and aggregation of prion proteins. Free Radical Biology and Medicine 2009, 46, 1353-1361, 10.1016/j.freeradbiomed.2009.02.013.
- Shouvik Roy; Bruce D. Mason; Christian S. Schöneich; John F. Carpenter; Thomas C. Boone; Bruce A. Kerwin; Light-induced aggregation of type I soluble tumor necrosis factor receptor. Journal of Pharmaceutical Sciences 2009, 98, 3182-3199, 10.1002/jps.21750.
- Shanghao Li; Roger M. Leblanc; Aggregation of Insulin at the Interface. The Journal of Physical Chemistry B 2013, 118, 1181-1188, 10.1021/jp4101202.
- Silvia Campioni; Guillaume Carret; Sophia Jordens; Lucrèce Nicoud; Raffaele Mezzenga; Roland Riek; The Presence of an Air–Water Interface Affects Formation and Elongation of α-Synuclein Fibrils. Journal of the American Chemical Society 2014, 136, 2866-2875, 10.1021/ja412105t.
- Ben J. Trigg; Chiu Fan Lee; David J. Vaux; Létitia Jean; The air–water interface determines the outcome of seeding during amyloidogenesis. Biochemical Journal 2013, 456, 67-80, 10.1042/bj20130605.
- Létitia Jean; Chiu Fan Lee; David J. Vaux; Enrichment of Amyloidogenesis at an Air-Water Interface. Biophysical Journal 2012, 102, 1154-1162, 10.1016/j.bpj.2012.01.041.
- Létitia Jean; Chiu Fan Lee; Chongsoo Lee; Michael Shaw; David J. Vaux; Competing discrete interfacial effects are critical for amyloidogenesis. The FASEB Journal 2009, 24, 309-317, 10.1096/fj.09-137653.
- Anna Pavlova; Chi-Yuan Cheng; Maia Kinnebrew; John Lew; Frederick W. Dahlquist; Songi Han; Protein structural and surface water rearrangement constitute major events in the earliest aggregation stages of tau. Proceedings of the National Academy of Sciences 2015, 113, E127-E136, 10.1073/pnas.1504415113.
- Thibaut Frachon; Franz Bruckert; Quentin Le Masne; Emmanuel Monnin; Marianne Weidenhaupt; Insulin Aggregation at a Dynamic Solid–Liquid–Air Triple Interface. Langmuir 2016, 32, 13009-13019, 10.1021/acs.langmuir.6b03314.
- V. Sluzky; J. A. Tamada; A. M. Klibanov; R. Langer; Kinetics of insulin aggregation in aqueous solutions upon agitation in the presence of hydrophobic surfaces.. Proceedings of the National Academy of Sciences 1991, 88, 9377-9381, 10.1073/pnas.88.21.9377.
- Mark Duerkop; Eva Berger; Astrid Dürauer; Alois Jungbauer; Impact of Cavitation, High Shear Stress and Air/Liquid Interfaces on Protein Aggregation. Biotechnology Journal 2018, 13, e1800062, 10.1002/biot.201800062.
- Murali Jayaraman; Patrick M. Buck; Arun Alphonse Ignatius; Kevin R. King; Wei Wang; Agitation-induced aggregation and subvisible particulate formation in model proteins. European Journal of Pharmaceutics and Biopharmaceutics 2014, 87, 299-309, 10.1016/j.ejpb.2014.01.004.
- Sylvia Kiese; Astrid Papppenberger; Wolfgang Friess; Hanns-Christian Mahler; Shaken, Not Stirred: Mechanical Stress Testing of an IgG1 Antibody. Journal of Pharmaceutical Sciences 2008, 97, 4347-4366, 10.1002/jps.21328.
- R. Matthew Fesinmeyer; Sabine Hogan; Atul Saluja; Stephen R. Brych; Eva Kras; Linda O. Narhi; David N. Brems; Yatin R. Gokarn; Effect of Ions on Agitation- and Temperature-Induced Aggregation Reactions of Antibodies. Pharmaceutical Research 2008, 26, 903-913, 10.1007/s11095-008-9792-z.
- Jun Zhang; Elizabeth M. Topp; Protein G, Protein A and Protein A-Derived Peptides Inhibit the Agitation Induced Aggregation of IgG. Molecular Pharmaceutics 2012, 9, 622-628, 10.1021/mp200548x.
- Renuka Thirumangalathu; Sampathkumar Krishnan; Margaret Speed Ricci; David N. Brems; Theodore W. Randolph; John F. Carpenter; Silicone Oil- and Agitation-Induced Aggregation of a Monoclonal Antibody in Aqueous Solution. Journal of Pharmaceutical Sciences 2009, 98, 3167-3181, 10.1002/jps.21719.
- Lotte Krielgaard; Latoya S. Jones; Theodore W. Randolph; Sven Frokjaer; James M. Flink; Mark C. Manning; John F. Carpenter; Effect of tween 20 on freeze-thawing- and agitation-induced aggregation of recombinant human factor XIII. Journal of Pharmaceutical Sciences 1998, 87, 1597-1603, 10.1021/js980126i.
- Alireza Abdolvahabi; Yunhua Shi; Sanaz Rasouli; Corbin M. Croom; Aleksandra Chuprin; Bryan F. Shaw; How Do Gyrating Beads Accelerate Amyloid Fibrillization?. Biophysical Journal 2017, 112, 250-264, 10.1016/j.bpj.2016.12.004.
- Lisa Kueltzo; W.E.I. Wang; Theodore W. Randolph; John F. Carpenter; Effects of Solution Conditions, Processing Parameters, and Container Materials on Aggregation of a Monoclonal Antibody during Freeze-Thawing. Journal of Pharmaceutical Sciences 2008, 97, 1801-1812, 10.1002/jps.21110.
- Tatiana Perevozchikova; Hirsh Nanda; Douglas P. Nesta; Christopher J. Roberts; Protein Adsorption, Desorption, and Aggregation Mediated by Solid-Liquid Interfaces. Journal of Pharmaceutical Sciences 2015, 104, 1946-1959, 10.1002/jps.24429.
- Alana Gerhardt; Nicole R. Mcgraw; Daniel Schwartz; Jared Bee; John F. Carpenter; Theodore W. Randolph; Protein Aggregation and Particle Formation in Prefilled Glass Syringes. Journal of Pharmaceutical Sciences 2014, 103, 1601-1612, 10.1002/jps.23973.
- Pinaki Basu; Sampathkumar Sampathkumarkrishnan; Renuka Thirumangalathu; Theodore W. Randolph; John F. Carpenter; IgG1 Aggregation and Particle Formation Induced by Silicone–water Interfaces on Siliconized Borosilicate Glass Beads: A Model for Siliconized Primary Containers. Journal of Pharmaceutical Sciences 2013, 102, 852-865, 10.1002/jps.23434.
- Innocent B. Bekard; Peter Asimakis; Joseph Bertolini; Dave E. Dunstan; The effects of shear flow on protein structure and function. Biopolymers 2011, 95, 733-745, 10.1002/bip.21646.
- Enhong Cao; Yahuei Chen; Zhanfeng Cui; Peter R. Foster; Effect of freezing and thawing rates on denaturation of proteins in aqueous solutions. Biotechnology and Bioengineering 2003, 82, 684-690, 10.1002/bit.10612.
- Yifat Miller; Buyong Ma; Ruth Nussinov; Zinc ions promote Alzheimer A aggregation via population shift of polymorphic states. Proceedings of the National Academy of Sciences 2010, 107, 9490-9495, 10.1073/pnas.0913114107.
- Kevin Pagel; Tomomi Seri; Hans von Berlepsch; Jan Griebel; Reinhard Kirmse; Christoph Böttcher; Beate Koksch; How Metal Ions Affect Amyloid Formation: Cu2+- and Zn2+-Sensitive Peptides. ChemBioChem 2008, 9, 531-536, 10.1002/cbic.200700656.
- Maria Hoernke; Dr. Beate Koksch; Dr. Gerald Brezesinski; Amyloidogenic Peptides at Hydrophobic-Hydrophilic Interfaces: Coordination Affinities and the Chelate Effect Dictate the Competitive Binding of Cu2+ and Zn2+. ChemPhysChem 2011, 12, 2225-2229, 10.1002/cphc.201100215.
- M. Hoernke; B. Koksch; G. Brezesinski; Influence of the hydrophobic interface and transition metal ions on the conformation of amyloidogenic model peptides. Biophysical Chemistry 2010, 150, 64-72, 10.1016/j.bpc.2010.02.014.
- Maria Hoernke; Jessica A. Falenski; Christian Schwieger; Beate Koksch; Gerald Brezesinski; Triggers for β-Sheet Formation at the Hydrophobic–Hydrophilic Interface: High Concentration, In-Plane Orientational Order, and Metal Ion Complexation. Langmuir 2011, 27, 14218-14231, 10.1021/la203016z.
- Jing Zhang; Xiang Y. Liu; Effect of protein–protein interactions on protein aggregation kinetics. The Journal of Chemical Physics 2003, 119, 10972, 10.1063/1.1622380.
- Celine Galvagnion; Alexander K Buell; Georg Meisl; Thomas Michaels; Michele Vendruscolo; Tuomas Knowles; Christopher M Dobson; Lipid vesicles trigger α-synuclein aggregation by stimulating primary nucleation. Nature Chemical Biology 2015, 11, 229-234, 10.1038/nchembio.1750.
- Galyna Gorbenko; Valeriya M. Ioffe; Paavo K.J. Kinnunen; Binding of Lysozyme to Phospholipid Bilayers: Evidence for Protein Aggregation upon Membrane Association. Biophysical Journal 2007, 93, 140-153, 10.1529/biophysj.106.102749.
- Evelyne Terzi; Günter Hölzemann; Joachim Seelig; Self-association of β-Amyloid Peptide (1–40) in Solution and Binding to Lipid Membranes. Journal of Molecular Biology 1995, 252, 633-642, 10.1006/jmbi.1995.0525.
- Hongxia Zhao; Esa K. J. Tuominen; Paavo K. J. Kinnunen; Formation of Amyloid Fibers Triggered by Phosphatidylserine-Containing Membranes. Biochemistry 2004, 43, 10302-10307, 10.1021/bi049002c.
- Emma Sparr; Maarten Engel; Dmitri V. Sakharov; Mariette Sprong; Jet Jacobs; Ben De Kruijff; Jo W.M. Höppener; J. Antoinette Killian; Islet amyloid polypeptide-induced membrane leakage involves uptake of lipids by forming amyloid fibers. FEBS Letters 2004, 577, 117-120, 10.1016/j.febslet.2004.09.075.
- Lv, Z.; Hashemi, M.; Banerjee, S.; Zagorski, K.; Rochet, J.-C.; Lyubchenko, Y.L. Phospholipid membranes promote the early stage assembly of α-synuclein aggregates. bioRxiv 2018, 295782.
- Abha Chauhan; Indrani Ray; Ved P. S. Chauhan; Interaction of amyloid beta-protein with anionic phospholipids: possible involvement of Lys28 and C-terminus aliphatic amino acids.. Neurochemical Research 2000, 25, 423-429, 10.1023/a:1007509608440.
- Canay Ege; Ka Yee C. Lee; Insertion of Alzheimer’s Aβ40 Peptide into Lipid Monolayers. Biophysical Journal 2004, 87, 1732-1740, 10.1529/biophysj.104.043265.
- Frank Ferrone; [17] Analysis of protein aggregation kinetics. Methods in Enzymology 1999, 309, 256-274, 10.1016/s0076-6879(99)09019-9.
- Aimee M. Morris; Murielle A. Watzky; Richard G. Finke; Protein aggregation kinetics, mechanism, and curve-fitting: A review of the literature. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2009, 1794, 375-397, 10.1016/j.bbapap.2008.10.016.
- Paolo Arosio; Tuomas P. J. Knowles; Sara Linse; On the lag phase in amyloid fibril formation. Physical Chemistry Chemical Physics 2015, 17, 7606-7618, 10.1039/c4cp05563b.
- James D. Harper; Peter T. Lansbury; MODELS OF AMYLOID SEEDING IN ALZHEIMER'S DISEASE AND SCRAPIE: Mechanistic Truths and Physiological Consequences of the Time-Dependent Solubility of Amyloid Proteins. Annual Review of Biochemistry 1997, 66, 385-407, 10.1146/annurev.biochem.66.1.385.
- Brian O'Nuallain; Shankaramma Shivaprasad; Indu Kheterpal; Ronald Wetzel; Thermodynamics of Aβ(1−40) Amyloid Fibril Elongation. Biochemistry 2005, 44, 12709-12718, 10.1021/bi050927h.
- Murielle A. Watzky; Aimee M. Morris; Eric D. Ross; Richard G. Finke; Fitting Yeast and Mammalian Prion Aggregation Kinetic Data with the Finke−Watzky Two-Step Model of Nucleation and Autocatalytic Growth†. Biochemistry 2008, 47, 10790-10800, 10.1021/bi800726m.
- ‡ Aimee M. Morris; Murielle Watzky; § And Jeffrey N. Agar; ‡ Richard G. Finke; Fitting Neurological Protein Aggregation Kinetic Data via a 2-Step, Minimal/“Ockham's Razor” Model: The Finke−Watzky Mechanism of Nucleation Followed by Autocatalytic Surface Growth. Biochemistry 2008, 47, 2413-2427, 10.1021/bi701899y.
- Murielle A. Watzky; Richard G. Finke; Transition Metal Nanocluster Formation Kinetic and Mechanistic Studies. A New Mechanism When Hydrogen Is the Reductant: Slow, Continuous Nucleation and Fast Autocatalytic Surface Growth. Journal of the American Chemical Society 1997, 119, 10382-10400, 10.1021/ja9705102.
- Bertrand Morel; Maria Paz Carrasco; Samuel Jurado; Carmen Marco; Francisco Conejero-Lara; Dynamic micellar oligomers of amyloid beta peptides play a crucial role in their aggregation mechanisms. Physical Chemistry Chemical Physics 2018, 20, 20597-20614, 10.1039/c8cp02685h.
- Jeremy D. Schmit; Kingshuk Ghosh; Ken Dill; What Drives Amyloid Molecules To Assemble into Oligomers and Fibrils?. Biophysical Journal 2011, 100, 450-458, 10.1016/j.bpj.2010.11.041.
- Filip Hasecke; Tatiana Miti; Carlos Perez; Jeremy Barton; Daniel Schölzel; Lothar Gremer; Clara S. R. Grüning; Garrett Matthews; Georg Meisl; Tuomas P. J. Knowles; et al.Dieter WillboldPhilipp NeudeckerHenrike HeiseGhanim UllahWolfgang HoyerMartin Muschol Origin of metastable oligomers and their effects on amyloid fibril self-assembly. Chemical Science 2018, 9, 5937-5948, 10.1039/c8sc01479e.
- Evan Powers; David L. Powers; The Kinetics of Nucleated Polymerizations at High Concentrations: Amyloid Fibril Formation Near and Above the “Supercritical Concentration”. Biophysical Journal 2006, 91, 122-132, 10.1529/biophysj.105.073767.
- Tricia R. Serio; Anil G. Cashikar; Anthony S. Kowal; George J. Sawicki; Jahan J. Moslehi; Louise Serpell; Morton F. Arnsdorf; Susan L. Lindquist; Nucleated Conformational Conversion and the Replication of Conformational Information by a Prion Determinant. Science 2000, 289, 1317-1321, 10.1126/science.289.5483.1317.
- Ziao Fu; Darryl Aucoin; Judianne Davis; William E. Van Nostrand; Steven O. Smith; Mechanism of Nucleated Conformational Conversion of Aβ42. Biochemistry 2015, 54, 4197-4207, 10.1021/acs.biochem.5b00467.
- Jiyong Lee; Elizabeth K. Culyba; Evan Powers; Jeffery W. Kelly; Amyloid-β forms fibrils by nucleated conformational conversion of oligomers. Nature Chemical Biology 2011, 7, 602-609, 10.1038/nchembio.624.
- Sandra Chimon; Medhat A Shaibat; Christopher R Jones; Diana C Calero; Buzulagu Aizezi; Yoshitaka Ishii; Evidence of fibril-like β-sheet structures in a neurotoxic amyloid intermediate of Alzheimer's β-amyloid. Nature Structural & Molecular Biology 2007, 14, 1157-1164, 10.1038/nsmb1345.
- David Ruzafa; Bertrand Morel; Lorena Varela; Ana I. Azuaga; Francisco Conejero-Lara; Characterization of Oligomers of Heterogeneous Size as Precursors of Amyloid Fibril Nucleation of an SH3 Domain: An Experimental Kinetics Study. PLOS ONE 2012, 7, e49690, 10.1371/journal.pone.0049690.
- David Ruzafa; Francisco Conejero-Lara; Bertrand Morel; Modulation of the stability of amyloidogenic precursors by anion binding strongly influences the rate of amyloid nucleation. Physical Chemistry Chemical Physics 2013, 15, 15508-15517, 10.1039/c3cp52313f.
- Bertrand Morel; David Ruzafa; Francisco Conejero-Lara; SH3 Domains as Suitable Models to Study Amyloid Aggregation. SH Domains 2015, 15(37), 1-15, 10.1007/978-3-319-20098-9_1.
- Nicolas Fay; Yuji Inoue; Luc Bousset; Hideki Taguchi; Ronald Melki; Assembly of the Yeast Prion Ure2p into Protein Fibrils. Journal of Biological Chemistry 2003, 278, 30199-30205, 10.1074/jbc.m303000200.
- A. M. Bhattacharyya; A. K. Thakur; R. Wetzel; Polyglutamine aggregation nucleation: Thermodynamics of a highly unfavorable protein folding reaction. Proceedings of the National Academy of Sciences 2005, 102, 15400-15405, 10.1073/pnas.0501651102.
- Avanish S. Parmar; Paul E. Gottschall; Martin Muschol; Pre-assembled clusters distort crystal nucleation kinetics in supersaturated lysozyme solutions. Biophysical Chemistry 2007, 129, 224-234, 10.1016/j.bpc.2007.06.002.
- Shannon E. Hill; Joshua Robinson; Garrett Matthews; Martin Muschol; Amyloid Protofibrils of Lysozyme Nucleate and Grow Via Oligomer Fusion. Biophysical Journal 2009, 96, 3781-3790, 10.1016/j.bpj.2009.01.044.
- Joseph T. Jarrett; Peter T. Lansbury; Seeding “one-dimensional crystallization” of amyloid: A pathogenic mechanism in Alzheimer's disease and scrapie?. Cell 1993, 73, 1055-1058, 10.1016/0092-8674(93)90635-4.
- S. I. A. Cohen; S. Linse; L. M. Luheshi; E. Hellstrand; D. A. White; L. Rajah; Daniel Otzen; Michele Vendruscolo; C. M. Dobson; T. P. J. Knowles; et al. Proliferation of amyloid- 42 aggregates occurs through a secondary nucleation mechanism. Proceedings of the National Academy of Sciences 2013, 110, 9758-9763, 10.1073/pnas.1218402110.
- Georg Meisl; Xiaoting Yang; Christopher M. Dobson; Sara Linse; Tuomas P. J. Knowles; Modulation of electrostatic interactions to reveal a reaction network unifying the aggregation behaviour of the Aβ42 peptide and its variants. Chemical Science 2017, 8, 4352-4362, 10.1039/c7sc00215g.
- Samuel I. A. Cohen; Michele Vendruscolo; Mark E. Welland; Christopher M. Dobson; Eugene M. Terentjev; Tuomas P. J. Knowles; Nucleated polymerization with secondary pathways. I. Time evolution of the principal moments. The Journal of Chemical Physics 2011, 135, 065105-065105, 10.1063/1.3608916.
- Samuel I. A. Cohen; Michele Vendruscolo; Christopher M. Dobson; Tuomas P. J. Knowles; Nucleated polymerization with secondary pathways. II. Determination of self-consistent solutions to growth processes described by non-linear master equations. The Journal of Chemical Physics 2011, 135, 065106-065106, 10.1063/1.3608917.
- Tuomas P. J. Knowles; Christopher A. Waudby; Glyn L. Devlin; Samuel I. A. Cohen; Adriano Aguzzi; Michele Vendruscolo; Eugene M. Terentjev; Mark E. Welland; Christopher M. Dobson; An Analytical Solution to the Kinetics of Breakable Filament Assembly. Science 2009, 326, 1533-1537, 10.1126/science.1178250.
- Sara Linse; Monomer-dependent secondary nucleation in amyloid formation. Biophysical Reviews 2017, 9, 329-338, 10.1007/s12551-017-0289-z.
- Mattias Törnquist; Thomas C. T. Michaels; Kalyani Sanagavarapu; Xiaoting Yang; Georg Meisl; Samuel I. A. Cohen; Tuomas P. J. Knowles; Sara Linse; Secondary nucleation in amyloid formation. Chemical Communications 2018, 54, 8667-8684, 10.1039/c8cc02204f.
- Georg Meisl; Xiaoting Yang; Erik Hellstrand; Birgitta Frohm; Julius Kirkegaard; Samuel I. A. Cohen; Christopher M. Dobson; Sara Linse; Tuomas Knowles; Differences in nucleation behavior underlie the contrasting aggregation kinetics of the Aβ40 and Aβ42 peptides. Proceedings of the National Academy of Sciences 2014, 111, 9384-9389, 10.1073/pnas.1401564111.
- Gayathri Ramachandran; Jayant B. Udgaonkar; Evidence for the Existence of a Secondary Pathway for Fibril Growth during the Aggregation of Tau. Journal of Molecular Biology 2012, 421, 296-314, 10.1016/j.jmb.2012.01.007.
- Alexander K. Buell; Celine Galvagnion; Ricardo Gaspar; Emma Sparr; Michele Vendruscolo; Tuomas Knowles; Sara Linse; Christopher M. Dobson; Solution conditions determine the relative importance of nucleation and growth processes in -synuclein aggregation. Proceedings of the National Academy of Sciences 2014, 111, 7671-7676, 10.1073/pnas.1315346111.
- Ricardo Gaspar; Georg Meisl; Alexander K. Buell; Laurence Young; Clemens F. Kaminski; Tuomas P. J. Knowles; Emma Sparr; Sara Linse; Secondary nucleation of monomers on fibril surface dominatesα-synuclein aggregation and provides autocatalytic amyloid amplification. Quarterly Reviews of Biophysics 2017, 50, e6-e6, 10.1017/s0033583516000172.
- Shae Padrick; Andrew D. Miranker; Islet Amyloid: Phase Partitioning and Secondary Nucleation Are Central to the Mechanism of Fibrillogenesis. Biochemistry 2002, 41, 4694-4703, 10.1021/bi0160462.
- Vito Foderà; Fabio Librizzi; Minna Groenning; Marco van de Weert; Maurizio Leone; Secondary Nucleation and Accessible Surface in Insulin Amyloid Fibril Formation. The Journal of Physical Chemistry B 2008, 112, 3853-3858, 10.1021/jp710131u.
- Dushyant Kumar Garg; Bishwajit Kundu; Clues for divergent, polymorphic amyloidogenesis through dissection of amyloid forming steps of bovine carbonic anhydrase and its critical amyloid forming stretch. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2016, 1864, 794-804, 10.1016/j.bbapap.2016.03.019.
- Samuel I. A. Cohen; Michele Vendruscolo; Christopher M. Dobson; Tuomas P. J. Knowles; Nucleated Polymerisation in the Presence of Pre-Formed Seed Filaments. International Journal of Molecular Sciences 2011, 12, 5844-5852, 10.3390/ijms12095844.
- Rodrigo Morales; Ines Moreno-Gonzalez; Claudio Soto; Cross-Seeding of Misfolded Proteins: Implications for Etiology and Pathogenesis of Protein Misfolding Diseases. PLOS Pathogens 2013, 9, e1003537, 10.1371/journal.ppat.1003537.
- Lary C. Walker; Marc I. Diamond; Karen E. Duff; Bradley T. Hyman; Mechanisms of Protein Seeding in Neurodegenerative Diseases. JAMA Neurology 2013, 70, 304-310, 10.1001/jamaneurol.2013.1453.
- J. H. Come; P. E. Fraser; P. T. Lansbury; A kinetic model for amyloid formation in the prion diseases: importance of seeding.. Proceedings of the National Academy of Sciences 1993, 90, 5959-5963, 10.1073/pnas.90.13.5959.
- Patrik Brundin; Ronald Melki; Ron R Kopito; Prion-like transmission of protein aggregates in neurodegenerative diseases. Nature Reviews Molecular Cell Biology 2010, 11, 301-307, 10.1038/nrm2873.
- Lary C. Walker; Mathias Jucker; Neurodegenerative Diseases: Expanding the Prion Concept. Annual Review of Neuroscience 2015, 38, 87-103, 10.1146/annurev-neuro-071714-033828.
- Mathias Jucker; Lary C. Walker; Pathogenic protein seeding in alzheimer disease and other neurodegenerative disorders. Annals of Neurology 2011, 70, 532-540, 10.1002/ana.22615.
- Kenjiro Ono; Ryoichi Takahashi; Tokuhei Ikeda; Masahito Yamada; Cross-seeding effects of amyloid β-protein and α-synuclein. Journal of Neurochemistry 2012, 122, 883-890, 10.1111/j.1471-4159.2012.07847.x.
- Fumio Oosawa; Michiki Kasai; A theory of linear and helical aggregations of macromolecules. Journal of Molecular Biology 1962, 4, 10-21, 10.1016/s0022-2836(62)80112-0.
- Oosawa, F.; Asakura, S. Thermodynamics of the Polymerization of Protein; Academic Press: London, UK, 1975.
- David Ruzafa; Lorena Varela; Ana I. Azuaga; Francisco Conejero-Lara; Bertrand Morel; Mapping the structure of amyloid nucleation precursors by protein engineering kinetic analysis. Physical Chemistry Chemical Physics 2013, 16, 2989-3000, 10.1039/c3cp54383h.
- Yukiko Hori; Tadafumi Hashimoto; Yosuke Wakutani; Katsuya Urakami; Kenji Nakashima; Margaret M. Condron; Satoshi Tsubuki; Takaomi C. Saido; David B. Teplow; Takeshi Iwatsubo; et al. The Tottori (D7N) and English (H6R) Familial Alzheimer Disease Mutations Accelerate Aβ Fibril Formation without Increasing Protofibril Formation. Journal of Biological Chemistry 2007, 282, 4916-4923, 10.1074/jbc.m608220200.
- Santosh Kumar; Jayant B. Udgaonkar; Conformational Conversion May Precede or Follow Aggregate Elongation on Alternative Pathways of Amyloid Protofibril Formation. Journal of Molecular Biology 2009, 385, 1266-1276, 10.1016/j.jmb.2008.11.033.
- Amy R. Hurshman; Joleen T. White; Evan T. Powers; Jeffery W. Kelly; Transthyretin Aggregation under Partially Denaturing Conditions Is a Downhill Polymerization. Biochemistry 2004, 43, 7365-7381, 10.1021/bi049621l.
- Tiago Q. Faria; Zaida L. Almeida; Pedro F. Cruz; Catarina S. H. Jesus; Pedro Castanheira; Rui M. M. Brito; A look into amyloid formation by transthyretin: aggregation pathway and a novel kinetic model. Physical Chemistry Chemical Physics 2015, 17, 7255-7263, 10.1039/c4cp04549a.
- Brian O'nuallain; Darragh B. Freir; Andrew J. Nicoll; Emmanuel Risse; Neil Ferguson; Caroline E. Herron; John Collinge; Dominic M. Walsh; Amyloid -Protein Dimers Rapidly Form Stable Synaptotoxic Protofibrils. The Journal of Neuroscience 2010, 30, 14411-14419, 10.1523/jneurosci.3537-10.2010.
- Vijayaraghavan Rangachari; Brenda D. Moore; Dana Kim Reed; Leilani K. Sonoda; Alexander W. Bridges; Erin Conboy; David Hartigan; Terrone L. Rosenberry; Amyloid-β(1−42) Rapidly Forms Protofibrils and Oligomers by Distinct Pathways in Low Concentrations of Sodium Dodecylsulfate. Biochemistry 2007, 46, 12451-12462, 10.1021/bi701213s.
- Gayathri Ramachandran; Jayant B. Udgaonkar; Understanding the Kinetic Roles of the Inducer Heparin and of Rod-like Protofibrils during Amyloid Fibril Formation by Tau Protein. Journal of Biological Chemistry 2011, 286, 38948-38959, 10.1074/jbc.m111.271874.
- Josué Juárez; Pablo Taboada; Victor Mosquera; Existence of Different Structural Intermediates on the Fibrillation Pathway of Human Serum Albumin. Biophysical Journal 2009, 96, 2353-2370, 10.1016/j.bpj.2008.12.3901.
- Nikolaj K. Holm; Stine K. Jespersen; Lise V. Thomassen; Tine Y. Wolff; Pankaj Sehgal; Line A. Thomsen; Gunna Christiansen; Christian Beyschau Andersen; Anders D. Knudsen; Daniel Otzen; et al. Aggregation and fibrillation of bovine serum albumin. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2007, 1774, 1128-1138, 10.1016/j.bbapap.2007.06.008.
- Silvia Campioni; Maria Francesca Mossuto; Silvia Torrassa; Giulia Calloni; Patrizia Polverino de Laureto; Annalisa Relini; Angelo Fontana; Fabrizio Chiti; Conformational properties of the aggregation precursor state of HypF-N. Journal of Molecular Biology 2008, 379, 554-567, 10.1016/j.jmb.2008.04.002.
- Patrizia Marinelli; Susanna Navarro; Manuel Bano Polo; Bertrand Morel; Ricardo Graña-Montes; Anna Sabé; Francesc Canals; Maria Rosario Fernandez; Francisco Conejero Lara; Salvador Ventura; et al. Global Protein Stabilization Does Not Suffice to Prevent Amyloid Fibril Formation. ACS Chemical Biology 2018, 13, 2094-2105, 10.1021/acschembio.8b00607.
- Martino Calamai; Niccolo Taddei; Massimo Stefani; Giampietro Ramponi; Fabrizio Chiti; Relative Influence of Hydrophobicity and Net Charge in the Aggregation of Two Homologous Proteins. Biochemistry 2003, 42, 15078-15083, 10.1021/bi030135s.
- Shuo Yang; Michael D.W. Griffin; Katrina J. Binger; Peter Schuck; Geoffrey J. Howlett; An Equilibrium Model for Linear and Closed-Loop Amyloid Fibril Formation. Journal of Molecular Biology 2012, 421, 364-377, 10.1016/j.jmb.2012.02.026.
- Michael D.W. Griffin; Melva L.Y. Mok; LeAnne M. Wilson; Chi L.L. Pham; Lynne J. Waddington; Matthew A. Perugini; Geoffrey J. Howlett; Phospholipid Interaction Induces Molecular-level Polymorphism in Apolipoprotein C-II Amyloid Fibrils via Alternative Assembly Pathways. Journal of Molecular Biology 2008, 375, 240-256, 10.1016/j.jmb.2007.10.038.
- Bertrand Morel; Lorena Varela; Ana I. Azuaga; Francisco Conejero-Lara; Environmental Conditions Affect the Kinetics of Nucleation of Amyloid Fibrils and Determine Their Morphology. Biophysical Journal 2010, 99, 3801-3810, 10.1016/j.bpj.2010.10.039.
- Jesús Zurdo; J. Iñaki Guijarro; Jose L Jiménez; Helen R Saibil; Christopher M Dobson; Dependence on solution conditions of aggregation and amyloid formation by an SH3 domain. Journal of Molecular Biology 2001, 311, 325-340, 10.1006/jmbi.2001.4858.
- Lorena Varela; Bertrand Morel; Ana I. Azuaga; Francisco Conejero-Lara; A single mutation in an SH3 domain increases amyloid aggregation by accelerating nucleation, but not by destabilizing thermodynamically the native state. FEBS Letters 2009, 583, 801-806, 10.1016/j.febslet.2009.01.033.
- Alexander L. Watters; Pritilekha Deka; Colin Corrent; David Callender; Gabriele Varani; Tobin Sosnick; David Baker; The Highly Cooperative Folding of Small Naturally Occurring Proteins Is Likely the Result of Natural Selection. Cell 2007, 128, 613-624, 10.1016/j.cell.2006.12.042.
- Michele Vendruscolo; E. Paci; M. Karplus; C. M. Dobson; Structures and relative free energies of partially folded states of proteins. Proceedings of the National Academy of Sciences 2003, 100, 14817-14821, 10.1073/pnas.2036516100.
- David Brockwell; Sheena E Radford; Intermediates: ubiquitous species on folding energy landscapes?. Current Opinion in Structural Biology 2007, 17, 30-37, 10.1016/j.sbi.2007.01.003.
- Thomas Jahn; Sheena E. Radford; Folding versus aggregation: Polypeptide conformations on competing pathways. Archives of Biochemistry and Biophysics 2008, 469, 100-117, 10.1016/j.abb.2007.05.015.
- Konstantin K. Turoverov; Irina M. Kuznetsova; Vladimir N. Uversky; The protein kingdom extended: Ordered and intrinsically disordered proteins, their folding, supramolecular complex formation, and aggregation. Progress in Biophysics and Molecular Biology 2010, 102, 73-84, 10.1016/j.pbiomolbio.2010.01.003.
- Yoshiki Sekijima; Luke Wiseman; Jeanne Matteson; Per Hammarstrom; Sean R. Miller; Anu R. Sawkar; William E. Balch; Jeffery W. Kelly; The Biological and Chemical Basis for Tissue-Selective Amyloid Disease. Cell 2005, 121, 73-85, 10.1016/j.cell.2005.01.018.
- Samuel I.A. Cohen; Michele Vendruscolo; Christopher M. Dobson; Tuomas Knowles; From Macroscopic Measurements to Microscopic Mechanisms of Protein Aggregation. Journal of Molecular Biology 2012, 421, 160-171, 10.1016/j.jmb.2012.02.031.
- Katy E. Routledge; Gian Gaetano Tartaglia; Geoffrey W. Platt; Michele Vendruscolo; Sheena E. Radford; Competition between Intramolecular and Intermolecular Interactions in an Amyloid-Forming Protein. Journal of Molecular Biology 2009, 389, 776-786, 10.1016/j.jmb.2009.04.042.
- A.J Modler; K Gast; G Lutsch; G Damaschun; Assembly of Amyloid Protofibrils via Critical Oligomers—A Novel Pathway of Amyloid Formation. Journal of Molecular Biology 2003, 325, 135-148, 10.1016/s0022-2836(02)01175-0.
- D. Thirumalai; G B Reddy; Are native proteins metastable?. Nature Chemistry 2011, 3, 910-911, 10.1038/nchem.1207.
- Fabrizio Chiti; Niccolò Taddei; Fabiana Baroni; Cristina Capanni; Massimo Stefani; Giampietro Ramponi; Christopher M. Dobson; Kinetic partitioning of protein folding and aggregation. Nature Genetics 2002, 9, 137-143, 10.1038/nsb752.
- José L. Jiménez; Ewan J. Nettleton; Mario Bouchard; Carol Robinson; Christopher M. Dobson; Helen R. Saibil; The protofilament structure of insulin amyloid fibrils. Proceedings of the National Academy of Sciences 2002, 99, 9196-9201, 10.1073/pnas.142459399.
- Timo Eichner; Sheena Radford; A Diversity of Assembly Mechanisms of a Generic Amyloid Fold. Molecular Cell 2011, 43, 8-18, 10.1016/j.molcel.2011.05.012.
- Andrew J. Baldwin; Tuomas P. J. Knowles; Gian Gaetano Tartaglia; Anthony W. Fitzpatrick; Glyn L. Devlin; Sarah Lucy Shammas; Christopher A. Waudby; Maria F. Mossuto; Sarah Meehan; Sally L. Gras; et al.John ChristodoulouSpencer J. Anthony-CahillPaul D. BarkerMichele VendruscoloChristopher M. Dobson Metastability of Native Proteins and the Phenomenon of Amyloid Formation. Journal of the American Chemical Society 2011, 133, 14160-14163, 10.1021/ja2017703.
- Song-Ho Chong; Sihyun Ham; Distinct Role of Hydration Water in Protein Misfolding and Aggregation Revealed by Fluctuating Thermodynamics Analysis. Accounts of Chemical Research 2015, 48, 956-965, 10.1021/acs.accounts.5b00032.
- Margaret Sunde; Colin C. F. Blake; From the globular to the fibrous state: protein structure and structural conversion in amyloid formation. Quarterly Reviews of Biophysics 1998, 31, 1-39, 10.1017/s0033583598003400.
- Stephanie M Dorta-Estremera; Jingjing Li; Wei Cao; Rapid Generation of Amyloid from Native Proteins In vitro. Journal of Visualized Experiments 2013, 82, e50869-e50869, 10.3791/50869.
- Lukasz Goldschmidt; Poh K. Teng; Roland Riek; David Eisenberg; Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proceedings of the National Academy of Sciences 2010, 107, 3487-3492, 10.1073/pnas.0915166107.
- Ronald Wetzel; Kinetics and Thermodynamics of Amyloid Fibril Assembly. Accounts of Chemical Research 2006, 39, 671-679, 10.1021/ar050069h.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.9K
Revision:
1 time
(View History)
Update Date:
23 Dec 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No