 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yo Niida | + 3508 word(s) | 3508 | 2021-12-08 18:11:00 | | | |
| 2 | Conner Chen | Meta information modification | 3508 | 2021-12-17 03:11:17 | | |
Video Upload Options
Human hereditary malformation syndromes are caused by mutations in the genes of the signal transduction molecules involved in fetal development. Among them, the Sonic hedgehog (SHH) signaling pathway is the most important, and many syndromes result from its disruption. The output of the SHH pathway is shown as GLI activity, which is generated by SHH in a concentration-dependent manner, i.e., the sum of activating form of GLI (GLIA) and repressive form of GLI (GLIR). Which gene is mutated and whether the mutation is loss-of-function or gain-of-function determine in which concentration range of SHH the imbalance occurs. In human malformation syndromes, too much or too little GLI activity produces symmetric phenotypes affecting brain size, craniofacial (midface) dysmorphism, and orientation of polydactyly with respect to the axis of the limb.
1. The SHH Pathway
1.1. SHH Acts by Establishing a Morphogen Gradient
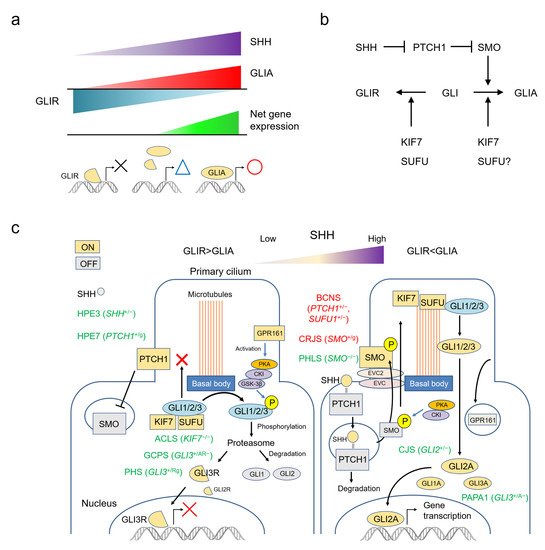
1.2. The SHH Pathway and The Primary Cilium
2. Human Malformation Syndromes Caused by Major Genes in the SHH Pathway
| Gene OMIM# | *165230 | *165240 | *600725 | *601309 | *601500 | *607035 | *611254 | ||||||||
| Gene | GLI2 | GLI3 | SHH | PTCH1 | SMO | SUFU | KIF7 | ||||||||
| Location | 2q14.2 | 7p14.1 | 7q36.3 | 9q22.32 | 7q32.1 | 10q24.32 | 15q26.1 | ||||||||
| Phenotype OMIM# | #615849 | #610829 | #175700 | #174200 | #146510 | #142945 | #109400 | #610828 | #241800 | #601707 | #109400 | #617757 | #200990 | #607131 | #614120 |
| Disease | CJS | HPE9 | GCPS | PAPA1 | PHS | HPE3 | BCNS | HPE7 | PHLS | CRJS | BCNS | JBTS32 | ACLS | AGBK | HLS2 |
| Inheritance | AD | AD | AD | AD | AD | AD | AD | AD | AR | Mos | AD | AR | AR | AR | AR |
| Mutation type | LOF | LOF | LOF 1 | LOF | GOF | LOF 2 | LOF | GOF | LOF | GOF | LOF 3 | partial LOF | LOF | LOF | LOF |
| Height | Short | Short | Short | Short | Tall | Short | Normal | ||||||||
| hypopituitalism | Y | Y | Y | Y | Y | ||||||||||
| Head size | Micro | Micro | Macro | Micro | Macro | Macro | Micro | Macro | Macro | Macro | Macro | ||||
| Holoprosencephaly | less common | variable degree | less common | variable degree | variable degree | Anencephaly | |||||||||
| Intellectual disability | some patients | Y | Normal, mild (rare) | Y | less common | Y | Speech delay | mild to moderate | less common | mild | Severe | Y | |||
| Eyes (telorism) | Hypo | Hypo | Hyper | Normal | Hypo | Hypo | Hyper | Hypo | Hyper | Hyper | Hyper | Hyper | |||
| Microphthalmia | Y | Y | Synophthalmia (in some) | Y | |||||||||||
| Mid-Face | hypo | hypo | wide | hypo | wide | hypo | wide | wide | |||||||
| Cleft lip/palate | both | both | both | both | both (5%) | both | Cleft palate | both (5%) | both | Cleft palate | |||||
| SMMCI | Y | Y | Y | ||||||||||||
| Hands Polydactyly | Post-Ax (some) | Post-Ax | Post/Pre-Ax | Post/Pre-Ax | Post-Ax | Post-Ax | Pre-Ax | Post-Ax | Post/Pre-Ax | Post-Ax | |||||
| Hand Syndactyly | Y | Y | Y | ||||||||||||
| Feet polydactyly | Post-Ax (in some) | Post-Ax | Post/Pre-Ax | Post/Pre-Ax | Post-Ax | Post-Ax | Pre-Ax (in some) | Post-Ax | Post/Pre-Ax | Post/Pre-Ax | |||||
| Feet syndactyly | Y | Y | Y | ||||||||||||
| Tumor | HTH | NBCC/DMB/OC | HTH | DMB | NBCC/DMB | ||||||||||
2.1. GLI2 (GLI Family Zinc Finger 2) *165230
2.1.1. Culler-Jones Syndrome; CJS #615849
2.1.2. Holoprosencephaly 9; HPE9 #610829
2.2. GLI3 (GLI Family Zinc Finger 3) *165240
2.2.1. Greig Cephalopolysyndactyly Syndrome; GCPS #175700
2.2.2. Polydactyly, Preaxial, Type IV; PPD4 #174700
2.2.3. Polydactyly, Postaxial, Types A1 and B; PAPA1 #174200
2.2.4. Pallister-Hall Syndrome; PHS #146510
2.3. SHH (Sonic Hedgehog Signalling Molecule) *600725
2.3.1. Holoprosencephaly 3; HPE3 #142945
2.3.2. Microphthalmia, Isolated, with Coloboma 5; MCOPCB5 #611638
2.3.3. Single Median Maxillary Central Incisor; SMMCI #147250 (SMMCI Syndrome Included)
2.4. PTCH1 (Patched 1) *601309
2.4.1. Basal Cell Nevus Syndrome; BCNS #109400
2.4.2. Holoprosencephaly 7; HPE7 #610828
2.5. SMO (Smoothened, Frizzled Class Receptor) *601500
2.5.1. Pallister-Hall-Like Syndrome; PHLS #241800
2.5.2. Curry-Jones Syndrome; CRJS #601707
References
- Hui, C.C.; Angers, S. Gli proteins in development and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 513–537.
- Shimeld, S.M.; van den Heuvel, M.; Dawber, R.; Briscoe, J. An amphioxus Gli gene reveals conservation of midline patterning and the evolution of hedgehog signalling diversity in chordates. PLoS ONE 2007, 2, e864.
- Ruppert, J.M.; Kinzler, K.W.; Wong, A.J.; Bigner, S.H.; Kao, F.T.; Law, M.L.; Seuanez, H.N.; O’Brien, S.J.; Vogelstein, B. The GLI-Kruppel family of human genes. Mol. Cell Biol. 1988, 8, 3104–3113.
- Villavicencio, E.H.; Walterhouse, D.O.; Iannaccone, P.M. The sonic hedgehog-patched-gli pathway in human development and disease. Am. J. Hum. Genet. 2000, 67, 1047–1054.
- Briscoe, J.; Thérond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429.
- Gorojankina, T. Hedgehog signaling pathway: A novel model and molecular mechanisms of signal transduction. Cell Mol. Life Sci. 2016, 73, 1317–1332.
- Kong, J.H.; Siebold, C.; Rohatgi, R. Biochemical mechanisms of vertebrate hedgehog signaling. Development 2019, 146, 6892.
- Sasai, N.; Toriyama, M.; Kondo, T. Hedgehog Signal and Genetic Disorders. Front. Genet. 2019, 10, 1103.
- Satir, P.; Pedersen, L.B.; Christensen, S.T. The primary cilium at a glance. J. Cell Sci. 2010, 123, 499–503.
- França, M.M.; Jorge, A.A.; Carvalho, L.R.; Costalonga, E.F.; Vasques, G.A.; Leite, C.C.; Mendonca, B.B.; Arnhold, I.J. Novel heterozygous nonsense GLI2 mutations in patients with hypopituitarism and ectopic posterior pituitary lobe without holoprosencephaly. J. Clin. Endocrinol. Metab. 2010, 95, E384–E391.
- Bertolacini, C.D.; Ribeiro-Bicudo, L.A.; Petrin, A.; Richieri-Costa, A.; Murray, J.C. Clinical findings in patients with GLI2 mutations—Phenotypic variability. Clin. Genet. 2012, 81, 70–75.
- Roessler, E.; Du, Y.Z.; Mullor, J.L.; Casas, E.; Allen, W.P.; Gillessen-Kaesbach, G.; Roeder, E.R.; Ming, J.E.; Ruiz i Altaba, A.; Muenke, M. Loss-of-function mutations in the human GLI2 gene are associated with pituitary anomalies and holoprosencephaly-like features. Proc. Natl. Acad. Sci. USA 2003, 100, 13424–13429.
- Niida, Y.; Inoue, M.; Ozaki, M.; Takase, E. Human Malformation Syndromes of Defective GLI: Opposite Phenotypes of 2q14.2 (GLI2) and 7p14.2 (GLI3) Microdeletions and a GLIA/R Balance Model. Cytogenet. Genome Res. 2017, 153, 56–65.
- Gustavsson, P.; Schoumans, J.; Staaf, J.; Jönsson, G.; Carlsson, F.; Kristoffersson, U.; Borg, A.; Nordenskjöld, M.; Dahl, N. Hemizygosity for chromosome 2q14.2-q22.1 spanning the GLI2 and PROC genes associated with growth hormone deficiency, polydactyly, deep vein thrombosis and urogenital abnormalities. Clin. Genet. 2006, 69, 441–443.
- Kordaß, U.; Schröder, C.; Elbracht, M.; Soellner, L.; Eggermann, T. A familial GLI2 deletion (2q14.2) not associated with the holoprosencephaly syndrome phenotype. Am. J. Med. Genet. A 2015, 167A, 1121–1124.
- Arnhold, I.J.; França, M.M.; Carvalho, L.R.; Mendonca, B.B.; Jorge, A.A. Role of GLI2 in hypopituitarism phenotype. J. Mol. Endocrinol. 2015, 54, R141–R150.
- Greally, M.T.; Robinson, E.; Allen, N.M.; O’Donovan, D.; Crolla, J.A. De novo interstitial deletion 2q14.1q22.1: Is there a recognizable phenotype? Am. J. Med. Genet. A 2014, 164A, 3194–3202.
- Kevelam, S.H.; van Harssel, J.J.; van der Zwaag, B.; Smeets, H.J.; Paulussen, A.D.; Lichtenbelt, K.D. A patient with a mild holoprosencephaly spectrum phenotype and heterotaxy and a 1.3 Mb deletion encompassing GLI2. Am. J. Med. Genet. A 2012, 158A, 166–173.
- Al-Qattan, M.M.; Shamseldin, H.E.; Salih, M.A.; Alkuraya, F.S. GLI3-related polydactyly: A review. Clin. Genet. 2017.
- Biesecker, L.G. The Greig cephalopolysyndactyly syndrome. Orphanet J. Rare Dis. 2008, 3, 10.
- Kroisel, P.M.; Petek, E.; Wagner, K. Phenotype of five patients with Greig syndrome and microdeletion of 7p13. Am. J. Med. Genet. 2001, 102, 243–249.
- Johnston, J.J.; Olivos-Glander, I.; Killoran, C.; Elson, E.; Turner, J.T.; Peters, K.F.; Abbott, M.H.; Aughton, D.J.; Aylsworth, A.S.; Bamshad, M.J.; et al. Molecular and clinical analyses of Greig cephalopolysyndactyly and Pallister-Hall syndromes: Robust phenotype prediction from the type and position of GLI3 mutations. Am. J. Hum. Genet. 2005, 76, 609–622.
- Fujioka, H.; Ariga, T.; Horiuchi, K.; Otsu, M.; Igawa, H.; Kawashima, K.; Yamamoto, Y.; Sugihara, T.; Sakiyama, Y. Molecular analysis of non-syndromic preaxial polydactyly: Preaxial polydactyly type-IV and preaxial polydactyly type-I. Clin. Genet. 2005, 67, 429–433.
- Biesecker, L.G.; Johnston, J. Syndromic and non-syndromic GLI3 phenotypes. Clin. Genet. 2005, 68, 285.
- Johnston, J.J.; Sapp, J.C.; Turner, J.T.; Amor, D.; Aftimos, S.; Aleck, K.A.; Bocian, M.; Bodurtha, J.N.; Cox, G.F.; Curry, C.J.; et al. Molecular analysis expands the spectrum of phenotypes associated with GLI3 mutations. Hum. Mutat. 2010, 31, 1142–1154.
- Nanni, L.; Ming, J.E.; Bocian, M.; Steinhaus, K.; Bianchi, D.W.; Die-Smulders, C.; Giannotti, A.; Imaizumi, K.; Jones, K.L.; Campo, M.D.; et al. The mutational spectrum of the sonic hedgehog gene in holoprosencephaly: SHH mutations cause a significant proportion of autosomal dominant holoprosencephaly. Hum. Mol. Genet. 1999, 8, 2479–2488.
- Schimmenti, L.A.; de la Cruz, J.; Lewis, R.A.; Karkera, J.D.; Manligas, G.S.; Roessler, E.; Muenke, M. Novel mutation in sonic hedgehog in non-syndromic colobomatous microphthalmia. Am. J. Med. Genet. A 2003, 116A, 215–221.
- Nanni, L.; Ming, J.E.; Du, Y.; Hall, R.K.; Aldred, M.; Bankier, A.; Muenke, M. SHH mutation is associated with solitary median maxillary central incisor: A study of 13 patients and review of the literature. Am. J. Med. Genet. 2001, 102, 1–10.
- Marini, M.; Cusano, R.; De Biasio, P.; Caroli, F.; Lerone, M.; Silengo, M.; Ravazzolo, R.; Seri, M.; Camera, G. Previously undescribed nonsense mutation in SHH caused autosomal dominant holoprosencephaly with wide intrafamilial variability. Am. J. Med. Genet. A 2003, 117A, 112–115.
- Kimonis, V.E.; Goldstein, A.M.; Pastakia, B.; Yang, M.L.; Kase, R.; DiGiovanna, J.J.; Bale, A.E.; Bale, S.J. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am. J. Med. Genet. 1997, 69, 299–308.
- Koch, C.A.; Chrousos, G.P.; Chandra, R.; Evangelista, R.S.; Gilbert, J.C.; Nobuhara, K.; Zhuang, Z.; Vortmeyer, A.O. Two-hit model for tumorigenesis of nevoid basal cell carcinoma (Gorlin) syndrome-associated hepatic mesenchymal tumor. Am. J. Med. Genet. 2002, 109, 74–76.
- Tate, G.; Kishimoto, K.; Mitsuya, T. Biallelic disruption of the PTCH1 gene in multiple basal cell carcinomas in Japanese patients with nevoid basal cell carcinoma syndrome. Acta. Med. Okayama 2014, 68, 163–170.
- Akizawa, Y.; Miyashita, T.; Sasaki, R.; Nagata, R.; Aoki, R.; Ishitani, K.; Nagashima, Y.; Matsui, H.; Saito, K. Gorlin syndrome with an ovarian leiomyoma associated with a PTCH1 second hit. Am. J. Med. Genet. A 2016, 170A, 1029–1034.
- Smyth, I.; Narang, M.A.; Evans, T.; Heimann, C.; Nakamura, Y.; Chenevix-Trench, G.; Pietsch, T.; Wicking, C.; Wainwright, B.J. Isolation and characterization of human patched 2 (PTCH2), a putative tumour suppressor gene inbasal cell carcinoma and medulloblastoma on chromosome 1p32. Hum. Mol. Genet. 1999, 8, 291–297.
- Metzis, V.; Courtney, A.D.; Kerr, M.C.; Ferguson, C.; Rondón Galeano, M.C.; Parton, R.G.; Wainwright, B.J.; Wicking, C. Patched1 is required in neural crest cells for the prevention of orofacial clefts. Hum. Mol. Genet. 2013, 22, 5026–5035.
- Abramyan, J. Hedgehog Signaling and Embryonic Craniofacial Disorders. J. Dev. Biol. 2019, 7, 9.
- Ming, J.E.; Kaupas, M.E.; Roessler, E.; Brunner, H.G.; Golabi, M.; Tekin, M.; Stratton, R.F.; Sujansky, E.; Bale, S.J.; Muenke, M. Mutations in PATCHED-1, the receptor for SONIC HEDGEHOG, are associated with holoprosencephaly. Hum. Genet. 2002, 110, 297–301.
- Ribeiro, L.A.; Murray, J.C.; Richieri-Costa, A. PTCH mutations in four Brazilian patients with holoprosencephaly and in one with holoprosencephaly-like features and normal MRI. Am. J. Med. Genet. A 2006, 140, 2584–2586.
- Derwińska, K.; Smyk, M.; Cooper, M.L.; Bader, P.; Cheung, S.W.; Stankiewicz, P. PTCH1 duplication in a family with microcephaly and mild developmental delay. Eur. J. Hum. Genet. 2009, 17, 267–271.
- Rubino, S.; Qian, J.; Pinheiro-Neto, C.D.; Kenning, T.J.; Adamo, M.A. A familial syndrome of hypothalamic hamartomas, polydactyly, and SMO mutations: A clinical report of 2 cases. J. Neurosurg. Pediatr. 2018, 23, 98–103.
- Le, T.L.; Sribudiani, Y.; Dong, X.; Huber, C.; Kois, C.; Baujat, G.; Gordon, C.T.; Mayne, V.; Galmiche, L.; Serre, V.; et al. Bi-allelic Variations of SMO in Humans Cause a Broad Spectrum of Developmental Anomalies Due to Abnormal Hedgehog Signaling. Am. J. Hum. Genet. 2020, 106, 779–792.
- Twigg, S.R.F.; Hufnagel, R.B.; Miller, K.A.; Zhou, Y.; McGowan, S.J.; Taylor, J.; Craft, J.; Taylor, J.C.; Santoro, S.L.; Huang, T.; et al. A Recurrent Mosaic Mutation in SMO, Encoding the Hedgehog Signal Transducer Smoothened, Is the Major Cause of Curry-Jones Syndrome. Am. J. Hum. Genet. 2016, 98, 1256–1265.

