Human hereditary malformation syndromes are caused by mutations in the genes of the signal transduction molecules involved in fetal development. Among them, the Sonic hedgehog (SHH) signaling pathway is the most important, and many syndromes result from its disruption. The output of the SHH pathway is shown as GLI activity, which is generated by SHH in a concentration-dependent manner, i.e., the sum of activating form of GLI (GLIA) and repressive form of GLI (GLIR). Which gene is mutated and whether the mutation is loss-of-function or gain-of-function determine in which concentration range of SHH the imbalance occurs. In human malformation syndromes, too much or too little GLI activity produces symmetric phenotypes affecting brain size, craniofacial (midface) dysmorphism, and orientation of polydactyly with respect to the axis of the limb.
1. The SHH Pathway
1.1. SHH Acts by Establishing a Morphogen Gradient
SHH is one of the most important morphogens in animals, and is involved in the pattern formation of many organs, including limbs and the ventral midline structure of the central nervous system [
25]. GLI family zinc finger proteins are the only known transcriptional effectors of the sonic hedgehog (SHH) signalling pathway; they control human embryonic development by regulating transcription of a group of target genes [
25]. The vertebrate
Gli gene family is thought to be derived from duplications of a single ancestral chordate gene [
26], and four
GLI genes—
GLI1 (12q13.3),
GLI2 (2q14.2),
GLI3 (7p14.1) and
GLI4 (8q24.3)—have been identified in humans [
27,
28]. GLI acts as a transcriptional enhancer (GLIA) in the presence of SHH, but suppresses the same target genes in the absence of SHH (GLIR). The output of the SHH pathway is expressed as a balance between GLIA and GLIR. The higher the SHH concentration, the higher the GLIA production; the lower the SHH concentration, the higher the GLIR production. The resulting net expression of target genes fluctuates continuously, and does not obey an all-or-nothing rule (
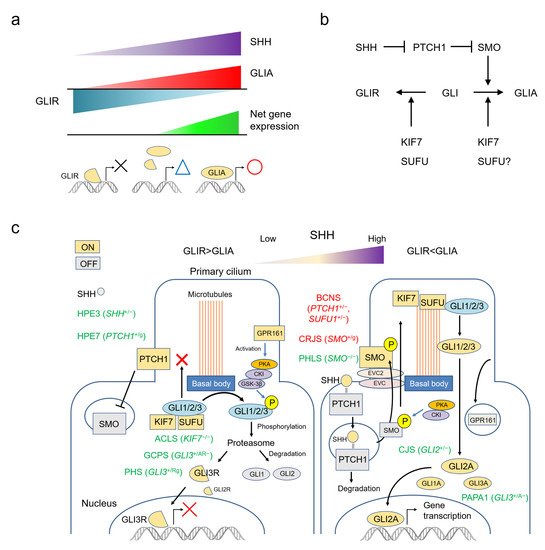
Figure 1a).
Figure 1. The SHH pathway and human malformation syndromes. (a) The output of the SHH pathway is expressed as a balance between GLIA and GLIR, fluctuating continuously rather than according to an all-or-nothing rule. (b) Simplified molecular circuit of the pathway. (c) Schematic representation of the relationship between the function of each molecule and the primary cilium, and associated syndromes. Each molecule changes its function by migrating to a specific site of the primary cilium. ACLS: acrocallosal syndrome; BCNS: basal cell nevus syndrome; CJS: Culler-Jones syndrome; CRJS: Curry-Jones syndrome; GCPS: Greig cephalopolysyndactyly syndrome; HPE: holoprosencephaly; PAPA1: polydactyly, postaxial, types A1 and B; PHLS: Pallister-Hall-like syndrome; PHS: Pallister-Hall syndrome.
1.2. The SHH Pathway and The Primary Cilium
Recently, the functions and mechanisms of the SHH pathway have been well-summarized in several reviews [
13,
25,
29,
30,
31]. To put it very simply based on these references, SMO is the key molecule that activates GLI into GLIA. PTCH1 constitutively suppresses the action of SMO, but this suppression is removed by SHH. As a result, SMO is activated in the presence of SHH and GLIA production is increased. Conversely, SMO remains suppressed in the absence of SHH, and GLI is converted to GLIR. KIF7 is involved in both activation and inhibition of GLI. SUFU mainly works to suppress GLI, but in some cases it may be involved in GLI activation (
Figure 1b).
The functions of molecules in the SHH pathway depend on the structure of the primary cilium (
Figure 1c). This solitary organelle emanates from the cell surface of most mammalian cell types during growth arrest. Increasing evidence suggests that primary cilia are key coordinators of signaling pathways—including PDGF, Hedgehog, Wnt and mechano-signaling—during development and in tissue homeostasis [
32].
The SHH signaling pathway can be summarized as follows: in the absence of SHH, the PTCH1 constitutively present in the primary cilium suppresses SMO. KIF7 and SUFU bind to the GLI protein, keeping it at the basal body and prohibiting entry of GLI into the cilium. Here, GLI is phosphorylated by the PKA, CKI, GSK-3β complex activated by GPR161, and degraded by the proteasome. Under these conditions, GLI1 and GLI2 are almost completely degraded, but GLI3 is converted to GLI3R and functions as an inhibitory transcription factor. When SHH is present, it binds to PTCH1 and promotes its degradation. Freed of PTCH1-mediated suppression, SMO is phosphorylated and translocated to the primary cilium. SUFU-bound GLI protein aggregates on the cilium tip, where GLI is released by the action of KIF7. Free GLI is then activated intracellularly. GLI2A is thought to be the most important activator, with the role of GLI3A small and that of GLI1A even smaller (
Figure 1c) [
13,
25,
29,
30,
31].
2. Human Malformation Syndromes Caused by Major Genes in the SHH Pathway
“Loss of function” (LOF) refers to a mutation in which a gene product (protein) is not produced or does not function if it is produced. On the other hand, “gain of function” (GOF) refers to a mutation in which a gene product is produced that functions abnormally. It does not matter whether its function is the same as or vice versa of the function that a normal gene product gives to the SHH pathway. The OMIM registration number of each gene and disease are listed. Refer the OMIM FAQs (
https://omim.org/help/faq#1_3; accessed on 8 November 2021) for the meaning of the symbols before the numbers. Core clinical features of each syndrome are summarized in
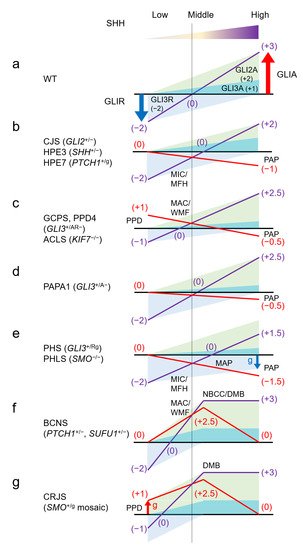
Table 1, and the effects of each gene mutation on GLIA/R balance are shown in
Figure 2. Many of these diseases are autosomal dominant, and fetal lethality is possible in those for which there are no reports of human genetic disorders due to homozygous LOF or GOF mutations. Similarly, some of these GOF mutations are only known to occur as somatic mosaics; in these cases, germline heterozygous mutations are also likely to cause embryonic lethality.
Figure 2. Human malformation syndromes and the GLIA/R balance model. Assume that suppression of GLI3R is −2 and activity of GLI2A and GLIA3 is 2 and 1, at maximum and minimum SHH concentrations, respectively. The output balance of GLIA/R is assumed to fluctuate linearly from zero to saturation of SHH concentration. The effect of the sum of GLIA and GLIR in each syndrome is shown by the purple line, and the deviation from the wild type (a) is shown by the red line. The actual amount ratio of GLIA and GLIR is unknown and may differ depending on the tissue, but it should be noted that the relationship between the direction of deviation of GLIA/R balance from the wild type and correlation to SHH concentration in each syndrome is unchanged. Preaxial polydactyly correlates with an overbalance in the low SHH region (thumb side) (c,g), and postaxial polydactyly correlates with an underbalance in the high SHH region (little finger side) (b–e). Microcephaly/midface hypoplasia correlates with an underbalance in the lower-intermediate SHH region (b,e), and macrocephaly/wide midface correlates with an overbalance (c,f). Also, neoplasia correlates with an overbalance in the intermediate SHH region (f,g). A: active form of GLI; AR: both active and repression form of GLI; ACLS: acrocallosal syndrome; BCNS: basal cell nevus syndrome; CJS: Culler-Jones syndrome; CRJS: Curry-Jones syndrome; DMB: desmoplastic medulloblastoma; g: gain of function mutation; GCPS: Greig cephalopolysyndactyly syndrome; HPE: holoprosencephaly; MAC: macrocephaly; MAP: mesoaxial polydactyly; MFH: midface hypoplasia; MIC: microcephaly; NBCC: nevoid basal cell carcinoma; PAP: postaxial polydactyly; PPD: preaxial polydactyly; PAPA1: polydactyly, postaxial, types A1 and B; PHLS: Pallister-Hall-like syndrome; PHS: Pallister-Hall syndrome; R: repression form of GLI;WMF: wide midface; WT: wild type.
Table 1. Clinical Synopsis in SHH Pathway syndromes.
| Gene OMIM# |
*165230 |
*165240 |
*600725 |
*601309 |
*601500 |
*607035 |
*611254 |
| Gene |
GLI2 |
GLI3 |
SHH |
PTCH1 |
SMO |
SUFU |
KIF7 |
| Location |
2q14.2 |
7p14.1 |
7q36.3 |
9q22.32 |
7q32.1 |
10q24.32 |
15q26.1 |
| Phenotype OMIM# |
#615849 |
#610829 |
#175700 |
#174200 |
#146510 |
#142945 |
#109400 |
#610828 |
#241800 |
#601707 |
#109400 |
#617757 |
#200990 |
#607131 |
#614120 |
| Disease |
CJS |
HPE9 |
GCPS |
PAPA1 |
PHS |
HPE3 |
BCNS |
HPE7 |
PHLS |
CRJS |
BCNS |
JBTS32 |
ACLS |
AGBK |
HLS2 |
| Inheritance |
AD |
AD |
AD |
AD |
AD |
AD |
AD |
AD |
AR |
Mos |
AD |
AR |
AR |
AR |
AR |
| Mutation type |
LOF |
LOF |
LOF 1 |
LOF |
GOF |
LOF 2 |
LOF |
GOF |
LOF |
GOF |
LOF 3 |
partial LOF |
LOF |
LOF |
LOF |
| Height |
Short |
Short |
|
|
Short |
|
|
|
Short |
|
|
Tall |
Short |
Normal |
|
| hypopituitalism |
Y |
Y |
|
|
Y |
Y |
|
Y |
|
|
|
|
|
|
|
| Head size |
Micro |
Micro |
Macro |
|
|
Micro |
Macro |
Macro |
Micro |
|
Macro |
Macro |
Macro |
Macro |
|
| Holoprosencephaly |
less common |
variable degree |
|
|
less common |
variable degree |
|
variable degree |
|
|
|
|
|
|
Anencephaly |
| Intellectual disability |
some patients |
Y |
Normal, mild (rare) |
|
|
Y |
less common |
Y |
Speech delay |
mild to moderate |
less common |
mild |
Severe |
Y |
|
| Eyes (telorism) |
Hypo |
Hypo |
Hyper |
Normal |
Hypo |
Hypo |
Hyper |
Hypo |
|
|
Hyper |
Hyper |
Hyper |
Hyper |
|
| Microphthalmia |
|
Y |
|
|
Y |
Synophthalmia (in some) |
|
|
|
Y |
|
|
|
|
|
| Mid-Face |
hypo |
hypo |
wide |
|
|
hypo |
wide |
hypo |
|
|
wide |
|
wide |
|
|
| Cleft lip/palate |
both |
both |
|
|
both |
both |
both (5%) |
both |
Cleft palate |
|
both (5%) |
|
both |
|
Cleft palate |
| SMMCI |
|
Y |
|
|
|
Y |
|
Y |
|
|
|
|
|
|
|
| Hands Polydactyly |
Post-Ax (some) |
Post-Ax |
Post/Pre-Ax |
Post/Pre-Ax |
Post-Ax |
|
|
|
Post-Ax |
Pre-Ax |
|
Post-Ax |
Post/Pre-Ax |
|
Post-Ax |
| Hand Syndactyly |
|
|
Y |
Y |
Y |
|
|
|
|
|
|
|
|
|
|
| Feet polydactyly |
Post-Ax (in some) |
Post-Ax |
Post/Pre-Ax |
Post/Pre-Ax |
Post-Ax |
|
|
|
Post-Ax |
Pre-Ax (in some) |
|
Post-Ax |
Post/Pre-Ax |
|
Post/Pre-Ax |
| Feet syndactyly |
|
|
Y |
|
Y |
|
|
|
Y |
|
|
|
|
|
|
| Tumor |
|
|
|
|
HTH |
|
NBCC/DMB/OC |
|
HTH |
DMB |
NBCC/DMB |
|
|
|
|
2.1. GLI2 (GLI Family Zinc Finger 2) *165230
2.1.1. Culler-Jones Syndrome; CJS #615849
CJS is caused by heterozygous LOF mutations in
GLI2. This autosomal dominant disorder is characterized by hypoplasia of the midface and the pituitary gland—which is in turn associated with anterior pituitary dysfunction—and/or postaxial polydactyly (PAP). The disorder shows incomplete penetrance and variable expressivity: some patients have midline facial defects and developmental delays [
63,
64,
65]. The patient manifests short stature due to growth hormone deficiency, and occasionally, a delay of puberty. Some patients manifest polydactyly, mostly postaxial without syndactyly, and variable degrees of intellectual and socio-behavioural disabilities. Genotype-phenotype correlations do not appear to fit the typically expected pattern: missense mutations are not always milder than truncating mutations, and point mutations are not always milder than chromosomal microdeletions [
66]. Although patients with large chromosomal deletions tend to have complicating visceral malformations, such as congenital heart defects, heterotaxy, and urogenital defects, the phenotypic spectra of point mutations and chromosomal deletions generally overlap and can be difficult to distinguish. Intellectual disability in particular is highly unpredictable. Notably, a patient with a 20 Mb deletion had normal intelligence [
67], and occasionally, several microdeletions are inherited from parents with normal or minimal phenotypes [
68].
According to the GLIA/R output balance regulation mechanism of the SHH pathway shown in Figure 1c, GLIR is generated in an environment with a low SHH concentration, and GLIA is produced in an environment with a high SHH concentration (Figure 2a). Since the main component of GLIR is GLI3, GLI2 haploinsufficiency does not affect the regions where the SHH concentration is low. On the other hand, since the main component of GLIA is GLI2, haploinsufficiency of GLI2 significantly decreases GLIA in the regions where SHH concentration is high. Additionally, even in the regions where the SHH concentration is intermediate, GLIA activity is lower than normal (Figure 2b). This abnormal GLIA/R balance is corelated with the development of the above-mentioned malformations that occur in CJS.
2.1.2. Holoprosencephaly 9; HPE9 #610829
HPE9 and CJS belong to a continuous phenotypic spectrum, with the relatively mild form referred as CJS and the severe form known as HPE9. HPE9 is characterized by a wide phenotypic spectrum of brain developmental defects, with or without overt forebrain cleavage abnormalities (holoprosencephaly). To date, many case reports and reviews of human
GLI2 defects associated with HPE9 have been made [
69,
70,
71], and the actual phenotypic spectra of these syndromes have been clarified [
66].
2.2. GLI3 (GLI Family Zinc Finger 3) *165240
2.2.1. Greig Cephalopolysyndactyly Syndrome; GCPS #175700
GCPS is caused by heterozygous LOF mutations in
GLI3. This autosomal dominant disease is characterised by polysyndactyly, macrocephaly, and correlated facial dysmorphisms (frontal bossing, ocular hypertelorism, and down-slanted palpebral fissures). Typical polysyndactyly features are bilateral preaxial polysyndactyly (PPD) of the feet, broad thumb, or PPD with simple syndactyly of other digits of the hands. In addition, PAP in the hands may present [
72]. GCPS can result from point mutations in
GLI3 or by contiguous gene deletion syndrome of 7p13 (GCPS-CGS). Most point mutations result in intellectually normal patients and manifest with variable grades of polysyndactyly [
73]. In contrast, GCPS-CGS is always associated with moderate to severe mental retardation and a consistent PPD of the big toe [
74].
The genotype–phenotype correlations of
GLI3 point mutations are well-established [
72,
75]. The N-terminal part of the GLI3 protein contains the zinc finger domain (ZFD; amino acid residues, AA 462–645). Truncating mutations upstream or within the ZFD abolish DNA-binding activity and result in a total loss of protein function if translated. Typically, these mutations result in GCPS. Considering the GLIA/R balance, functional loss of one of the
GLI3 alleles halves the amount of GLIR produced in the regions where the SHH concentration is low. Consequently, GLI activity is higher than normal in these regions. GLI3A also decreases in the regions where SHH concentration is high, but the effect is minimal because the main component of GLIA is GLI2. In the regions with intermediate SHH concentration, GLI activity is also higher than normal (
Figure 2c). These perturbations in GLIA/R balance can explain why GCPS presents predominantly with preaxial polydactyly (though occasionally with postaxial) and macrocephaly.
2.2.2. Polydactyly, Preaxial, Type IV; PPD4 #174700
PPD4 and GCPS belong to a continuous phenotypic spectrum. The cranio-facial features of GCPS may be minimal or unclear in some patients, who can then be diagnosed with PPD4 if their syndrome is limited to preaxial polydactyly [
76,
77].
2.2.3. Polydactyly, Postaxial, Types A1 and B; PAPA1 #174200
Heterozygous LOF mutations in
GLI3 can cause isolated polydactyly. Mutation of the C-terminal part of the GLI3 protein, which contains the transactivating domains that mediate the activator function of the protein, result in a phenotype that varies from typical GCPS to PPD4, or PAPA1 [isolated PAP with well-formed (type A1) or rudimentary (type B) digit] [
75,
78]. Precise genotype-phenotype correlations in these cases have not been established. If the mutated protein is expressed, it can convert to GLI3R normally with low SHH levels, but GLI3A activity is decreased under high SHH concentrations. This type of mutation may manifest as PAPA1 (
Figure 3d).
2.2.4. Pallister-Hall Syndrome; PHS #146510
PHS is caused by heterozygous GOF mutations in GLI3. Truncating mutations in the middle part of the protein generate a ZFD-only version of GLI3 that does not include the transactivating domains 1 and 2 (TA2, AA 1044–1322; and TA1, AA 1376–1580) located on the C-terminal end of the protein. This mutant protein acts similarly to GLI3R, and it cannot be converted to GLI3A, leading to an extreme abundance of GLI3R activity and decoupling it from SHH signaling.
PHS is characterised by midface hypoplasia similar to or more severe than CJS, postaxial or mesoaxial (near the center of the axis) polydactyly, and other central structure abnormalities, including hypothalamic hamartoma, bifid epiglottis or laryngeal cleft, and pulmonary segmentation anomalies.
In PHS, one allele of GLI3 produces constantly GLI3R by mutation, so the GLIA/R balance is generally inclined toward GLIR. However, in regions where the SHH concentration is very low, there is no excess or deficiency compared to normal. Therefore, in PHS, symptoms related to insufficient GLI activity appear strongly, but symptoms due to excess GLI activity such as GCPS do not appear. For polydactyly, GLIA deficiency affects the more central region of the anterior-posterior axis, resulting in mesoaxial polydactyly, but preaxial polydactyly does not occur (Figure 2e).
2.3. SHH (Sonic Hedgehog Signalling Molecule) *600725
Mutations in the SHH gene itself also contribute to human malformation syndromes, as a series of related symptoms. All of these syndromes are caused by LOF mutations in one allele of SHH, and are classified as autosomal dominant due to haploinsufficiency.
2.3.1. Holoprosencephaly 3; HPE3 #142945
HPE3 presents with varying degrees of holoprosencephaly-, microcephaly- and midface hypoplasia-related symptoms. Nanni et al. presented a panel of 12 photographs illustrating the range of severity in holoprosencephaly resulting from mutations in the
SHH gene [
79].
2.3.2. Microphthalmia, Isolated, with Coloboma 5; MCOPCB5 #611638
Schimmenti et al. reported a family with a heterozygous 24 bp deletion in
SHH. Proband is an 8-month-old boy with bilateral microphthalmia with bilateral iris, and right chorioretinal and left uveoretinal coloboma (missing of any of eye structure). The boy had no stigmata of holoprosencephaly or other malformations. Incomplete expression of the
SHH mutation was observed, as his mother, who had unilateral iris and uveoretinal coloboma, and 3 unaffected family members, all carried the same deletion [
80].
2.3.3. Single Median Maxillary Central Incisor; SMMCI #147250 (SMMCI Syndrome Included)
Single median maxillary central incisor (SMMCI) is one of the symptoms associated with dysplasia of the median facial structure. It is often associated with the holoprosencephaly spectrum, but can also occur as an isolated malformation [
81,
82].
2.4. PTCH1 (Patched 1) *601309
2.4.1. Basal Cell Nevus Syndrome; BCNS #109400
Heterozygous LOF mutations in
PTCH1 cause basal cell nevus syndrome (BCNS), also known as Gorlin syndrome. BCNS is characterized by numerous basal cell cancers of the skin, keratocysts of the jaws, palmar and plantar pits, ovarian fibromas, medulloblastomas, and various malformations. Craniofacial manifestations include macrocephaly, broad facies, frontal bossing, hypertelorism and broad nasal root (
Figure 2a). It is also associated with calcification of the falx cerebri, rib and vertebral abnormalities, cleft lip or cleft palate, and cortical defects of bones, but not polydactyly or syndactyly [
59,
83]. Kimonis et al. tabulated major and minor BCNS criteria, and defined diagnosis of BCNS in terms of the presence of two major or one major and two minor criteria [
59]. Biallelic disruption of
PTCH1 (second hit mutation) is reported in basal cell carcinomas [
84] and ovarian leiomyomas [
85] in patients with BCNS. BCNS is also caused by heterozygous LOF mutations in
PTCH2 [
86] and
SUFU (see
Section 4.6.1).
PTCH1 is a receptor of SHH; the binding of PTCH1 to SHH de-represses SMO, leading to downstream activation of GLI. When the amount of protein is halved by loss of one PTCH1 allele, the amount of SHH required for maximal GLI activation is also halved compared to the wild type. Therefore, there is no difference from wild type in regions where SHH concentration is at minimum or maximum, but activation of GLI is strongly enhanced in regions where the amount of SHH is intermediate or relatively low, compared to wild type (Figure 2f). This explains the presentation of macrocephaly and wide midface and the absence of polydactyly, and suggests an association with the development of cancer.
Cleft lip/palate is also observed in some patients with BCNS. In mice, loss of
Ptch1 function in cranial neural crest cells (which relieves SMO inhibition and leads to constitutive activation of the HH pathway) has been shown to cause mid-facial expansion, which culminates in cleft lip as well [
87]. It is thought that the same mechanism applies in humans [
55]. Therefore, cleft lip/palate can be caused by both less or excess of SHH pathway activity.
2.4.2. Holoprosencephaly 7; HPE7 #610828
Heterozygous GOF mutations in PTCH1 cause HPE with varying degrees of symptoms (HPE7) [
88,
89]. Missense mutations that produce a PTCH1 protein with reduced SHH binding capacity or reduced capacity for signal transduction attenuate SHH pathway activation. Such mutations in one of the
PTCH1 alleles have the same effect as LOF mutations that halve functional SHH protein (HPE3) (
Figure 2b).
Also, mothers and children with microcephaly and developmental delay due to a ~360 Kb duplication on chromosome 9q22.32, which includes
PTCH1, have been reported [
90]. In this case,
PTCH1 becomes 3 copies and the SHH pathway is strongly suppressed.
2.5. SMO (Smoothened, Frizzled Class Receptor) *601500
2.5.1. Pallister-Hall-Like Syndrome; PHLS #241800
PHLS caused by homozygous LOF mutations of
SMO. This autosomal recessive disease is characterized by microcephaly, facial dysmorphism associated with midface hypoplasia, and postaxial polydactyly with variable expressivity. Patients also exhibit hypothalamic hamartoma, cardiac and skeletal anomalies, and Hirschsprung disease [
91,
92]. Because SMO is the main transducer of positive SHH signalling, loss of SMO results in a broad reduction in GLI activity in regions with intermediate to high concentration of SHH—much more severe than in
GLI2 or
SHH haploinsufficiency. The profile of GLIA/R balance is expected to be similar to that of PHS (
Figure 2e).
2.5.2. Curry-Jones Syndrome; CRJS #601707
CRJS caused by GOF mutations in
SMO with somatic mosaicism. Twigg et al. studied tissue samples from 10 unrelated patients with Curry-Jones syndrome, and identified somatic mosaicism for an identical missense mutation in
SMO in all cases: NM_005631.4:c.1234C>T p.(Leu412Phe) [
93]. Curry-Jones syndrome is a multisystem disorder characterized by patchy skin lesions (hypopigmented streaky lesions), polysyndactyly, diverse cerebral malformations, unicoronal craniosynostosis, iris colobomas, microphthalmia, and intestinal malrotation with myofibromas or hamartomas.
The p.Leu412Phe mutation is thought to constitutively activate SMO and explains the development of preaxial polydactyly and medulloblastoma in the GLIA/R balance model (Figure 2g). In addition, CRJS is accompanied by asymmetry of the skull and orbit due to mosaic mutation. Skin symptoms and colorectal symptoms not found in other SHH pathway syndromes are observed, which are also considered to be the result of GOF mosaic mutations in SMO.
This entry is adapted from the peer-reviewed paper 10.3390/ijms222313060