Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Allan Rod Merrill | + 2205 word(s) | 2205 | 2021-12-07 04:08:21 | | | |
| 2 | Rita Xu | Meta information modification | 2205 | 2021-12-16 03:07:45 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Merrill, A. A Structural Approach to Anti-Virulence. Encyclopedia. Available online: https://encyclopedia.pub/entry/17171 (accessed on 26 July 2026).
Merrill A. A Structural Approach to Anti-Virulence. Encyclopedia. Available at: https://encyclopedia.pub/entry/17171. Accessed July 26, 2026.
Merrill, Allan. "A Structural Approach to Anti-Virulence" Encyclopedia, https://encyclopedia.pub/entry/17171 (accessed July 26, 2026).
Merrill, A. (2021, December 15). A Structural Approach to Anti-Virulence. In Encyclopedia. https://encyclopedia.pub/entry/17171
Merrill, Allan. "A Structural Approach to Anti-Virulence." Encyclopedia. Web. 15 December, 2021.
Copy Citation
The anti-virulence strategy is designed to prevent bacterial virulence factors produced by pathogenic bacteria from initiating and sustaining an infection.
mono-ADP-ribosyltransferase toxins
bacterial toxins
protein crystallography
anti-virulence strategy
1. Introduction
Since their serendipitous discovery by Alexander Fleming, antibiotics have been the basis in modern medicine for the treatment of bacterial diseases because of the life-saving compounds produced by Florey during World War II. However, the emergence of multidrug resistance bacteria has threatened to unravel this approach to human and animal health. The global spread of resistant microbes in all areas of disease treatment has left the antibiotic drug pipeline unable to meet demand since compounds are metabolized by voracious multi-drug resistant organisms [1][2][3]. The situation begs for new strategies to combat microbial pathogens [2][4][5][6][7].
A compelling, innovative, and alternative approach to antibiotic therapy is the anti-virulence or anti-infective strategy that involves targeting virulence-associated rather than survival/fitness-relevant traits in the offending pathogen [8][9][10][11][12][13]. Thus, the “anti-infective” agent/drug interferes with the pathogen’s ability to cause disease [14][15]. One avenue for this approach is to target bacterial toxins using this powerful method [9][16][17][18][19]. This approach involves the use of patho- or virulence blockers developed specifically to bind bacterial virulence factors with high affinity, neutralizing or reducing the virulence of the offending pathogen [17][20][21]. Anti-virulence compounds offer significant advantages over conventional antibiotics. Firstly, these agents are directed towards specific mechanisms in the offending pathogen that promote infection or harm host cells rather than targeting an essential growth/metabolic factor. Disarming microorganisms of their virulence properties without threatening their survival offers lower selection pressure, reducing the risk of drug-resistant mutations. Secondly, virulence-specific therapeutics avoids the collateral damage on the host microbiota associated with current antibiotics. Other possible advantages to the anti-virulence approach include limited off-target effects due to the unique mechanism of the mART toxin action and targeting a conserved motif within a family of proteins provides a potential to target multiple pathogens with a single small molecule compound. Thus, the anti-virulence approach has the potential for a paradigm shift in the development and treatment of bacterial diseases and infections. This novel approach makes pathogenic bacteria less virulent by neutralizing their weaponry, thereby giving the host’s immune system time to clear the pathogen. Additionally, the weakened pathogen is more susceptible to antibiotic therapy [15].
Bacterial toxins of all types are a common infection tool produced by bacterial pathogens to cause disease. These factors facilitate the infection process by the pathogen and are often key elements in disease development. Compounds, known as anti-virulence agents, are designed to interfere or block the toxin machinery and associated molecular mechanisms and hold promise to thwart bacterial pathogens.
A growing collection of bacterial toxins that serve as high profile targets for anti-virulence intervention is the mono-ADP-ribosyltransferase (mART) family. Family members covalently modify specific host macromolecules as an “on—off” switch often producing a unique pathology. Notably, diphtheria toxin (DT) is a classic example of a mART toxin that is the sole cause of a disease (diphtheria) by modifying ribosomal elongation factor 2 (eEF2), terminating protein synthesis, and leading to apoptosis [22]. The cellular targets for mART toxins are often key regulators of cell function and include (i) GTPases, (ii) actin, (iii) kinase regulators, (iv) elongation factors, and (v) RNA-recognition motifs, and even DNA (target genes) [22][23][24][25][26][27][28]. We developed a data-mining strategy based on fold-recognition methods [29][30][31] that unearths new mARTs as targets for disease intervention using the anti-virulence or anti-infective approach (Figure 1).

Figure 1. Summary of mART toxin discovery pipeline. Step 1: Known mART toxin sequences are used (input) to provide several thousands (10,000–20,000) of candidate toxin sequences arising from iterative search tools such as HHblits and HHsenser. This mining strategy is analogous to a large-scale BLAST search that cater to protein homologs with low sequence identity [32][33]. Step 2: Sequences are then subjected to pattern-based filters, including searches for the conserved mART motif, followed by predictions for transmembrane domains (unwanted), and for secretion signal peptides or other indicators of non-classical secretion. Step 3: Finally, consensus fold recognition analyses determine whether the remaining sequences may yield a mART-like fold by generating homology models. The final list contains putative mART toxin sequences for experimental testing in a yeast growth-deficiency assay to validate computer predictions [34] [taken from Tremblay et al. Toxins 12: 792, 2020].
2. Anti-Virulence Approach
2.1. Virtual Screens
One facet of our approach is to use relevant high-resolution crystal structures as the template models for virtual screening (Figure 2). The PDB structure is prepared for molecular docking using appropriate software (OpenEye, http://www.openeye.net/products/software/ (accessed on 5 May 2017); Schrodinger software suite, https://www.schrodinger.com/ (accessed on 6 June 2018) run on a super-computer network [35][36]. Virtual screens yield a list of potential “hits” against the anti-virulence drug target (receptor) and can produce valuable contributions in “hit-and lead-compound” discovery [37][38][39] with promising results [40][41][42][43][44][45] using iota toxin in complex with NADH (1GIQ:C2-like) [46][47] and C3bot1 toxin (2C8A:C3-like) [48]. Online compound libraries such as the ZINC Drugs NOW set of 6.5 million drug-like compounds https://zinc12.docking.org/subsets/drugs-now (accessed on 6 June 2018) serve as a starting source of compounds for docking experiments with methods as described [48]. Ultimately, only a small, manageable subset of compounds (usually less than 100) was selected for testing as inhibitors of mART toxin enzyme and cytotoxic activity [46][48][47].
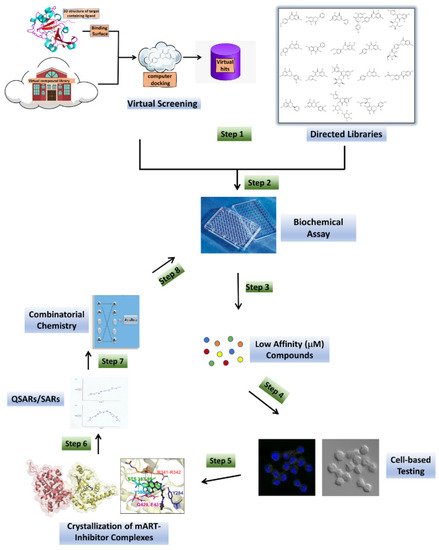
Figure 2. Anti-virulence development pipeline for mART toxins. mART inhibitors arise from two major sources, namely, virtual screening using a high-resolution mART structure of a toxin-inhibitor complex and “hand-picked” directed libraries (Step 1). Candidate compounds are then subjected to a Biochemical Assay (Step 2), producing a library of active, low-affinity compounds (μmol/L affinity for binding site), which are then chosen for cell-based testing (Step 3). Promising inhibitor compounds are then tested for efficacy to protect the corresponding target host cells from the mART toxin (Step 4). The most efficacious compounds (leads) are co-crystallized (Step 5) with their cognate toxin followed by lead optimization through Structure–Activity Relationships/Quantitative Structure–Activity Relationships methodology (Step 6). Combinatorial Chemistry methods are then used to prepare derivatives of the best compounds (Step 7), which are then re-tested in the Biochemical Assay (Step 8) and the cycle of development continues until higher-affinity compounds (nmol/L or greater) are developed.
2.2. Directed Libraries
We recently chose flavonoid compounds as our directed library source of small molecules [49] to complement the virtual screen approach. Flavonoids are natural products produced as plant secondary metabolites and are polyphenols that feature two phenyl rings bridged with a pyran or pyranone ring (see Table 1). Importantly, flavonoids show diverse biological activities, including antibacterial activity [50], inhibitors of proteins in cells, or their membranes and membrane perturbation activity [51][52]. Initially, a flavonoid library of 1200 compounds were sourced as potential anti-virulence agents against mARTs, and finally, 20 compounds were chosen for experimental testing based on several filter criteria, including literature reports of antimicrobial activity.
Table 1. Chemical Inhibitors of Plx2A.
| Inhibitor | Chemical Name | Structure | a KD (μmol/L) |
b IC50 (μmol/L) |
|---|---|---|---|---|
| Acacetin | 5,7-dihydroxy-2-(4-methoxy phenyl)-4H-chromen-4-one | 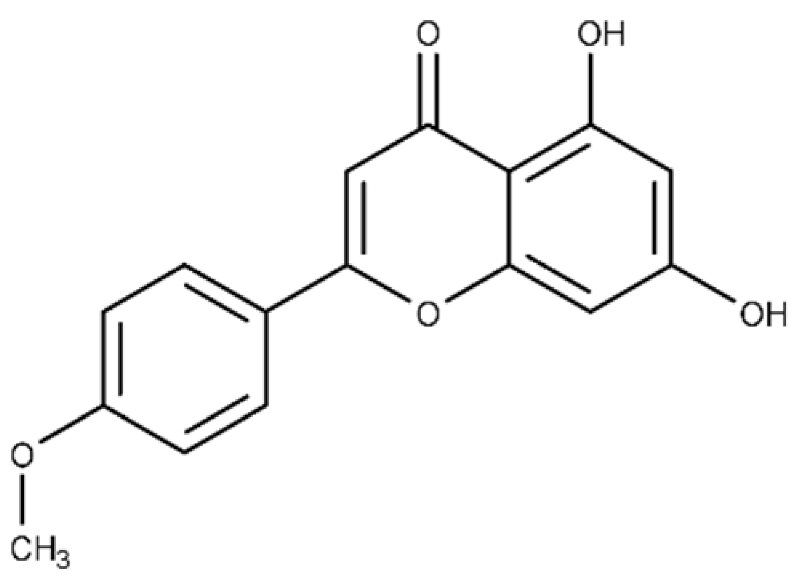 |
c ND | 28.0 ± 2.3 |
| Baicalein | 5,6,7-trihydroxy-2-phenyl-4H-chromen-4-one | 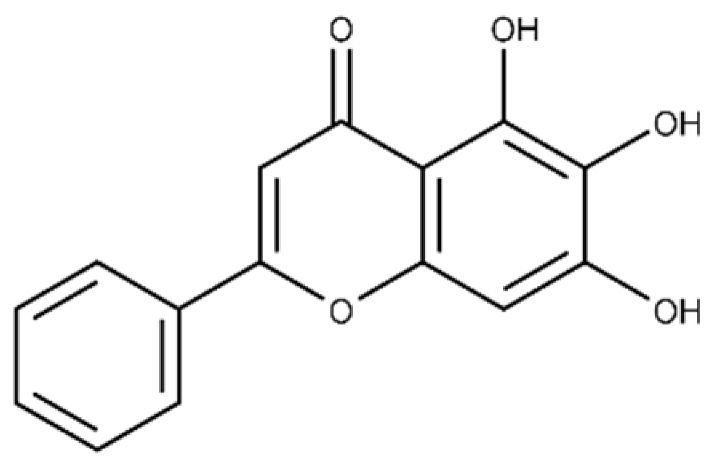 |
13.6 ± 1.5 | 10.7 ± 0.7 |
| Chrysin | 5,7-dihydroxy-2-phenyl-4H-chromen-4-one | 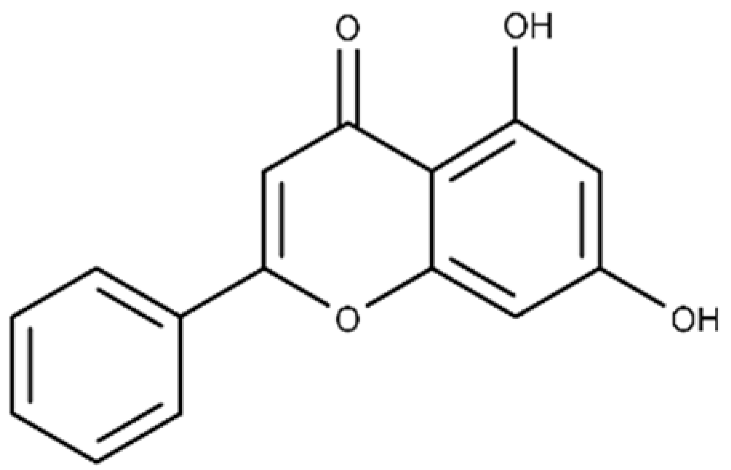 |
40.6 ± 4.7 | 49.2 ± 2.5 |
| Jaceosidin | 5,7-dihydroxy-2-(4-hydroxy-3-methoxyphenyl)-6-methoxy-4H-chromen-4-one | 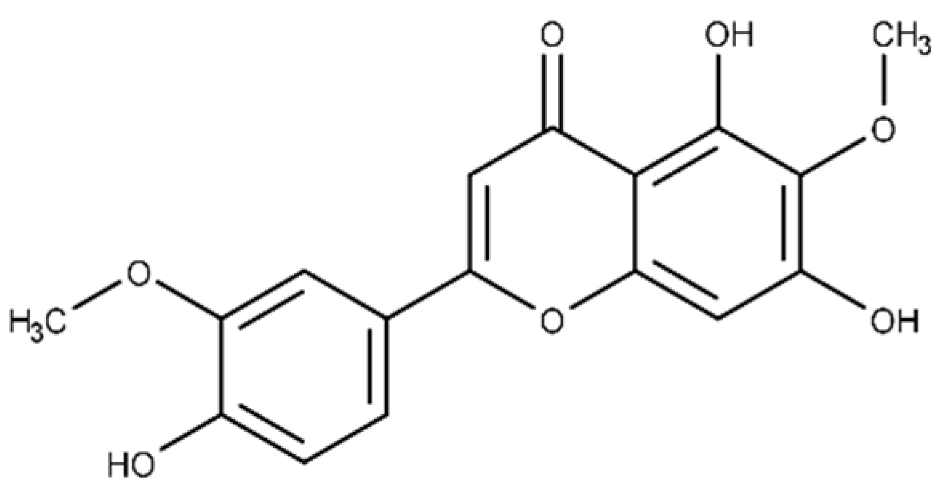 |
24.1 ± 2.9 | 73.9 ± 4.7 |
| Kaempferol | 3,5,7-trihydroxy-2-(4-hydroxyphenyl)-4H-chromen-4-one | 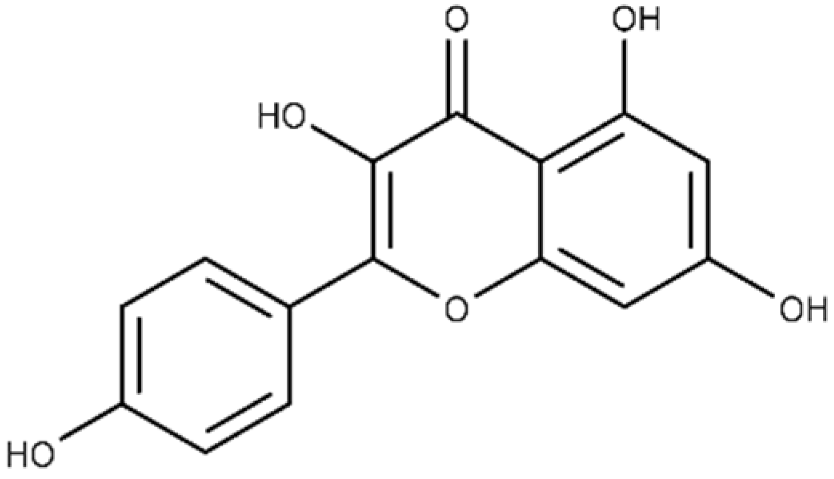 |
37.3 ± 3.6 | 79.9 ± 6.0 |
| Luteolin | 2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-4H-chromen-4-one | 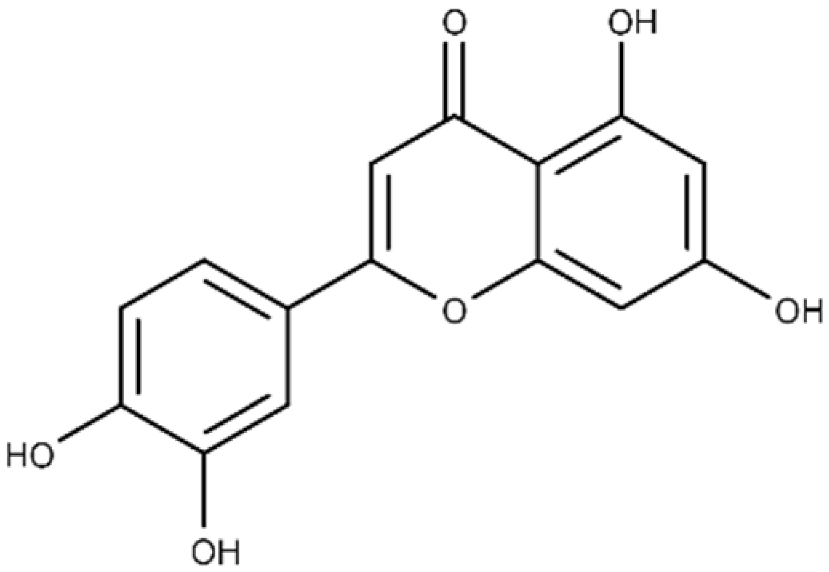 |
24.0 ± 1.5 | 68.1 ± 1.8 |
| Morin | 2-(2,4-dihydroxyphenyl)-3,5,7-trihydroxy-4H-chromen-4-one | 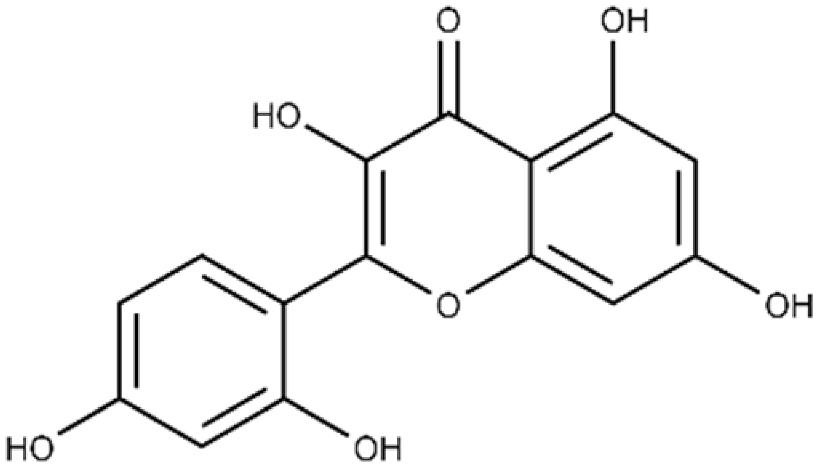 |
32.8 ± 11.7 | 109 ± 4.8 |
| Quercetin | 2-(3,4-dihydroxyphenyl)-3,5,7-trihydroxy-4H-chromen-4-one | 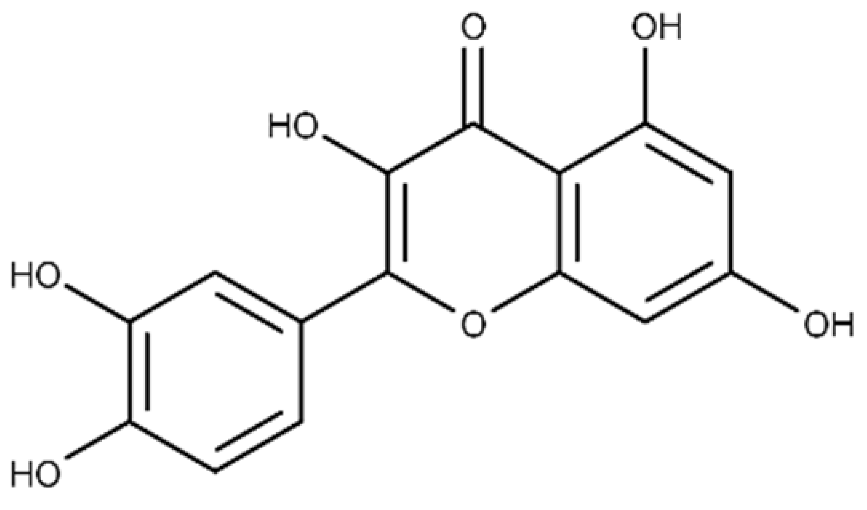 |
19.9 ± 0.4 | 24.3 ± 0.8 |
| M3 | N-{[(3R)-1-{1H-pyrazolo[3,4-d]pyrimidin-4- yl}piperidin-3-yl]methyl}methanesulfon amide |
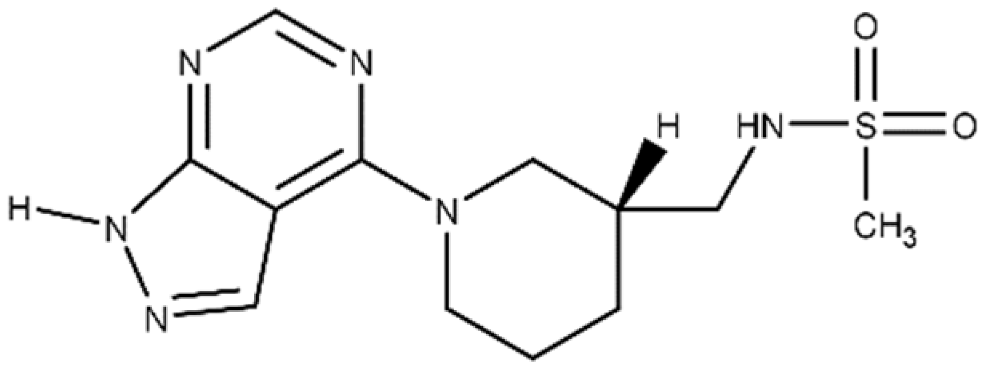 |
a The binding affinity of inhibitors to Plx2A (KD) were calculated from the binding isotherms generated from the quenching of intrinsic Trp fluorescence of the protein upon titration with the inhibitor (ligand). The values are the mean ± S.D. from three separate experiments. b The IC50 values were calculated from the fit to a dose–response curve of Plx2A GH activity against the concentration of inhibitor/flavonoid compounds to produce a directed library of 20 compounds for experimental testing against Plx2A activity [9]. c ND, not determined.
2.3. Biochemical Assay
Initial testing of the inhibitory activity against mART toxins involves an enzymatic assay, since these virulence factors are bacterial enzymes that cleave the NAD+ substrate and transfer ADP-ribose to the macromolecule substrate. However, most mARTs also show a secondary activity known as glycohydrolase (GH) that involves direct hydrolysis of NAD+ in the absence of a second substrate. A fluorescence-based plate-reader enzyme assay is often used to determine the inhibitor properties (IC50, Ki values) of the selected compounds (Figure 2). A fluorescence-based binding assay for lead compounds with the target mART is also established based on quenching of intrinsic fluorescence upon enzyme-inhibitor association to provide a measure of binding affinity (KD) [9].
2.4. Cell-Based Testing
Compounds with low micromolar (μmol/L) efficacy from the biochemical assay step (Figure 2) are then tested in a cell-based system that is specific for the mART-producing pathogen (Figure 2). Effective compounds are identified as those agents that provide protection of mART-intoxicated cells at low micromolar doses while not causing cellular cytotoxicity. This assay provides a direct measure of the dose–response (ED50 value) and cellular toxicity (TD50 value) of the inhibitor compounds. The best compounds are those with the highest therapeutic index (TD50/ED50) as a measure of inhibitor potential.
2.5. Crystallization of mART-Inhibitor Complexes
The co-crystal structures of mARTs with the best lead compounds are pursued to provide important structural biology insights into the location, nature, and chemistry of the inhibitor-binding sites (Figure 2). The details of the toxin-inhibitor complexes with the various mARTs provides new insights into the structural features/differences of the binding pockets for mART toxins. These structural data also provide the basis for further rationale inhibitor design to iteratively evolve/design lead hits into bona fide drug candidates.
2.6. SARs/QSARs and Combinatorial Chemistry
Rational inhibitor design and improvement involves SARs and QSARs studies to iteratively evolve lead hits into high-affinity drug candidates (Figure 2). Lead compounds identified in virtual screens or from “directed libraries” that show promise in enzyme and mammalian cell assays are further developed with respect to binding potency and pharmacokinetic properties. These compounds are investigated initially through a combination of modeling and docking studies using the target crystal structure to see which groups are essential for target binding, while at the same time introducing water-solubilizing groups with an eye to keeping the pKa values for compounds between 5–6. New second- and third-generation analogues can be synthesized using the unique microwave-assisted, continuous flow synthesis platform [53][54][55] (Figure 2). Conventional Lipinski parameters are used to guide analogue design [56], which are obtained using Drug Discovery software. As new candidates arise from the screen efforts, they are brought through the same optimization process and the cycle continues until high-affinity compounds are achieved.
3. Anti-Virulence Agents against American Foulbrood
3.1. Paenibacillus Larvae
Honey bee (Apis mellifera) beekeepers are faced with multiple challenges globally when maintaining bee colonies for pollination and honey production [57]. American Foulbrood is a bacterial disease that afflicts the brood and is responsible for huge economic losses in the apiary [58][59]. The disease is caused by Paenibacillus larvae, a Gram-positive, rod-shaped bacterium that forms spores that can remain viable for decades [60]. The ERIC genotypes of P. larvae strains are named after their enterobacterial repetitive intergenic consensus sequences and exist as five strains, where only two are currently environmentally active [61]. ERICI is found worldwide and is the slowest-killing phenotype, which helps to avoid the housekeeping of nurse bees, while ERICII is the most virulent species that occurs only in Europe and can infect an entire colony within a week [62]. Several virulence factors produced by P. larvae have been identified and their biology and role in pathogenicity have been reported [58][63]. These factors include two mART toxins, Plx1 and Plx2, shown to be important ERICI virulence factors [64]; Plx2A was recently characterized and crystallized [65]. However, to date, only a few of the P. larvae virulence factors have been targeted for an anti-virulence strategy [9][65][66].
3.2. C3larvin Toxin
C3larvin was identified by a bioinformatics approach as a mART in the C3 toxin subgroup and putative virulence factor from P. larvae. Cytoplasmic expression of C3larvin in a yeast model system revealed that it is highly cytotoxic [48]. C3larvin catalytic variants (Q155A, E157A, Q155A/E157A) with reduced or no mART enzymatic activity, were not toxic to yeast (eukaryotic model), revealing that C3larvin was a mART toxin with enzymatically driven cytotoxicity in a eukaryotic host. C3larvin showed mART enzyme activity and labeled Asn41 on RhoA, as seen in the C3 subgroup [48]. Surprisingly, C3larvin did not enter target macrophages unlike other C3 subgroup members, and it was determined that C3larvin possessed a truncated helix 1. It is now appreciated that it is only functional in a unique P. larvae strain of the MLST sequence type 9 in ERICIII/IV [66]. Two crystal structures of C3larvin revealed that it is indeed a C3 subgroup member with a shortened helix 1 (PDB:TR5; PDB:5DZQ), although the protein is not functional in bee larval assays [67].
An inhibitor, M3, of C3larvin mART enzymatic activity was identified by a virtual screen [47]. The methane sulfonamide derivative inhibited the enzyme with a Ki = 11 μmol/L and was the first inhibitor reported for the mART C3 subgroup [48]. M3 is unusual as an inhibitor of mART toxins because it possesses an adenine ring linked to substituted piperidine. M3 was also shown to be an effective inhibitor against Vis toxin from Vibrio splendidus and C3bot1 from Clostridium botulinum [47]. A molecular mechanics/molecular dynamics approach suggested that M3 competes with the adenine ring of the NAD+ substrate, but verification awaits a high-resolution co-crystal structure [48].
References
- Jian, Z.; Zeng, L.; Xu, T.; Sun, S.; Yan, S.; Yang, L.; Huang, Y.; Jia, J.; Dou, T. Antibiotic resistance genes in bacteria: Occurrence, spread, and control. J. Basic Microbiol. 2021, 62, 1049–1070.
- Rubey, K.M.; Brenner, J.S. Nanomedicine to fight infectious disease. Adv. Drug Deliv. Rev. 2021, 179, 113996.
- Tompson, A.C.; Manderson, L.; Chandler, C.I.R. Understanding antibiotic use: Practices, structures and networks. JAC Antimicrob. Resist. 2021, 3, dlab150.
- Mousavi, S.M.; Babakhani, S.; Moradi, L.; Karami, S.; Shahbandeh, M.; Mirshekar, M.; Mohebi, S.; Moghadam, M.T. Bacteriophage as a Novel Therapeutic Weapon for Killing Colistin-Resistant Multi-Drug-Resistant and Extensively Drug-Resistant Gram-Negative Bacteria. Curr. Microbiol. 2021, 78, 4023–4036.
- Bhardwaj, S.; Bhatia, S.; Singh, S.; Franco, F., Jr. Growing emergence of drug-resistant Pseudomonas aeruginosa and attenuation of its virulence using quorum sensing inhibitors: A critical review. Iran. J. Basic Med. Sci. 2021, 24, 699–719.
- Kang, D.; Zhang, L.; Kirienko, N.V. High-Throughput Approaches for the Identification of Pseudomonas aeruginosa Antivirulents. mBio 2021, 12, e02240-20.
- Cherier, D.; Patin, D.; Blanot, D.; Touze, T.; Barreteau, H. The Biology of Colicin M and Its Orthologs. Antibiotics 2021, 10, 1109.
- Hotinger, J.A.; Morris, S.T.; May, A.E. The Case against Antibiotics and for Anti-Virulence Therapeutics. Microorganisms 2021, 9, 2049.
- Ebeling, J.; Pieper, F.; Gobel, J.; Knispel, H.; McCarthy, M.; Goncalves, M.; Turner, M.; Merrill, A.R.; Genersch, E. Anti-Virulence Strategy against the Honey Bee Pathogenic Bacterium Paenibacillus larvae via Small Molecule Inhibitors of the Bacterial Toxin Plx2A. Toxins 2021, 13, 607.
- Ford, C.A.; Hurford, I.M.; Cassat, J.E. Antivirulence Strategies for the Treatment of Staphylococcus aureus Infections: A Mini Review. Front. Microbiol. 2020, 11, 632706.
- Lim, K.Y.L.; Mullally, C.A.; Haese, E.C.; Kibble, E.A.; McCluskey, N.R.; Mikucki, E.C.; Thai, V.C.; Stubbs, K.A.; Sarkar-Tyson, M.; Kahler, C.M. Anti-Virulence Therapeutic Approaches for Neisseria gonorrhoeae. Antibiotics 2021, 10, 103.
- Zhang, D.; Gan, R.Y.; Zhang, J.R.; Farha, A.K.; Li, H.B.; Zhu, F.; Wang, X.H.; Corke, H. Antivirulence properties and related mechanisms of spice essential oils: A comprehensive review. Compr. Rev. Food Sci. Food Saf. 2020, 19, 1018–1055.
- Stewart, D.; Anwar, F.; Vedantam, G. Anti-virulence strategies for Clostridioides difficile infection: Advances and roadblocks. Gut Microbes 2020, 12, 1802865.
- Yakovlieva, L.; Fulleborn, J.A.; Walvoort, M.T.C. Opportunities and Challenges of Bacterial Glycosylation for the Development of Novel Antibacterial Strategies. Front. Microbiol. 2021, 12, 745702.
- Lakemeyer, M.; Zhao, W.; Mandl, F.A.; Hammann, P.; Sieber, S.A. Thinking outside the box—Novel antibacterials to tackle the resistance crisis. Angew. Chem. Int. Ed. 2018, 57, 14440–14475.
- Lugo, M.R.; Merrill, A.R. Development of Anti-Virulence Therapeutics against Mono-ADP-Ribosyltransferase Toxins. Toxins 2020, 13, 16.
- Ahmad-Mansour, N.; Loubet, P.; Pouget, C.; Dunyach-Remy, C.; Sotto, A.; Lavigne, J.P.; Molle, V. Staphylococcus aureus Toxins: An Update on Their Pathogenic Properties and Potential Treatments. Toxins 2021, 13, 677.
- Hu, H.; Liu, M.; Sun, S. Pore-Forming Toxins During Bacterial Infection: Molecular Mechanisms and Potential Therapeutic Targets. Drug Des. Dev. Ther. 2021, 15, 3773–3781.
- Kim, B.S. Spatiotemporal Regulation of Vibrio Exotoxins by HlyU and Other Transcriptional Regulators. Toxins 2020, 12, 544.
- Vij, R.; Hube, B.; Brunke, S. Uncharted territories in the discovery of antifungal and antivirulence natural products from bacteria. Comput. Struct. Biotechnol. J. 2021, 19, 1244–1252.
- Hotinger, J.A.; Pendergrass, H.A.; May, A.E. Molecular Targets and Strategies for Inhibition of the Bacterial Type III Secretion System (T3SS); Inhibitors Directly Binding to T3SS Components. Biomolecules 2021, 11, 316.
- Collier, R.J. Understanding the mode of action of diphtheria toxin: A perspective on progress during the 20th century. Toxicon 2001, 39, 1793–1803.
- Lyons, B.; Ravulapalli, R.; Lanoue, J.; Lugo, M.R.; Dutta, D.; Carlin, S.; Merrill, A.R. Scabin, a Novel DNA-acting ADP-ribosyltransferase from Streptomyces scabies. J. Biol. Chem. 2016, 291, 11198–11215.
- Simon, N.C.; Aktories, K.; Barbieri, J.T. Novel bacterial ADP-ribosylating toxins: Structure and function. Nat. Rev. Microbiol. 2014, 12, 599–611.
- Grimaldi, G.; Corda, D.; Catara, G. From toxins to mammalian enzymes: The diversity of mono-ADP-ribosylation. Front. Biosci. Landmark Ed. 2015, 20, 389–404.
- Yoshida, T.; Tsuge, H. Common Mechanism for Target Specificity of Protein- and DNA-Targeting ADP-Ribosyltransferases. Toxins 2021, 13, 40.
- Aktories, K.; Lang, A.E.; Schwan, C.; Mannherz, H.G. Actin as target for modification by bacterial protein toxins. FEBS J. 2011, 278, 4526–4543.
- Vogelsgesang, M.; Pautsch, A.; Aktories, K. C3 exoenzymes, novel insights into structure and action of Rho-ADP-ribosylating toxins. Naunyn Schmiedeberg’s Arch. Pharmacol. 2007, 374, 347–360.
- Fieldhouse, R.J.; Turgeon, Z.; White, D.; Merrill, A.R. Cholera- and anthrax-like toxins are among several new ADP-ribosyltransferases. PLoS Comput. Biol. 2010, 6, e1001029.
- Fieldhouse, R.J.; Merrill, A.R. Needle in the haystack: Structure-based toxin discovery. Trends Biochem. Sci. 2008, 33, 546–556.
- Tremblay, O.; Thow, Z.; Geddes-McAlister, J.; Merrill, A.R. Several New Putative Bacterial ADP-Ribosyltransferase Toxins Are Revealed from In Silico Data Mining, Including the Novel Toxin Vorin, Encoded by the Fire Blight Pathogen, Erwinia amylovora. Toxins 2020, 12, 792.
- Remmert, M.; Biegert, A.; Hauser, A.; Soding, J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2011, 9, 173–175.
- Soding, J.; Remmert, M.; Biegert, A.; Lupas, A.N. HHsenser: Exhaustive transitive profile search using HMM-HMM comparison. Nucleic Acids Res. 2006, 34, W374–W378.
- Turgeon, Z.; White, D.; Jorgensen, R.; Visschedyk, D.; Fieldhouse, R.J.; Mangroo, D.; Merrill, A.R. Yeast as a tool for characterizing mono-ADP-ribosyltransferase toxins. FEMS Microbiol. Lett. 2009, 300, 97–106.
- Swann, S.L.; Brown, S.P.; Muchmore, S.W.; Patel, H.; Merta, P.; Locklear, J.; Hajduk, P.J. A unified, probabilistic framework for structure- and ligand-based virtual screening. J. Med. Chem. 2011, 54, 1223–1232.
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749.
- Schneider, G. Virtual screening: An endless staircase? Nat. Rev. Drug Discov. 2010, 9, 273–276.
- Rollinger, J.M.; Stuppner, H.; Langer, T. Virtual screening for the discovery of bioactive natural products. Prog. Drug Res. 2008, 65, 211, 213–249.
- Zhu, T.; Cao, S.; Su, P.C.; Patel, R.; Shah, D.; Chokshi, H.B.; Szukala, R.; Johnson, M.E.; Hevener, K.E. Hit identification and optimization in virtual screening: Practical recommendations based on a critical literature analysis. J. Med. Chem. 2013, 56, 6560–6572.
- Jorgensen, W.L. Efficient drug lead discovery and optimization. Acc. Chem. Res. 2009, 42, 724–733.
- Cerqueira, N.M.; Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Virtual screening of compound libraries. Methods Mol. Biol. 2009, 572, 57–70.
- Sousa, S.F.; Cerqueira, N.M.; Fernandes, P.A.; Ramos, M.J. Virtual screening in drug design and development. Comb. Chem. High Throughput Screen. 2010, 13, 442–453.
- Irwin, J.J.; Raushel, F.M.; Shoichet, B.K. Virtual screening against metalloenzymes for inhibitors and substrates. Biochemistry 2005, 44, 12316–12328.
- Danishuddin, M.; Khan, A.U. Structure based virtual screening to discover putative drug candidates: Necessary considerations and successful case studies. Methods 2015, 71, 135–145.
- Elseginy, S.A. Virtual screening and structure-based 3D pharmacophore approach to identify small-molecule inhibitors of SARS-CoV-2 Mpro. J. Biomol. Struct. Dyn. 2021, 1–17.
- Shniffer, A.; Visschedyk, D.D.; Ravulapalli, R.; Suarez, G.; Turgeon, Z.J.; Petrie, A.A.; Chopra, A.K.; Merrill, A.R. Characterization of an actin-targeting ADP-ribosyltransferase from Aeromonas hydrophila. J. Biol. Chem. 2012, 287, 37030–37041.
- Ravulapalli, R.; Lugo, M.R.; Pfoh, R.; Visschedyk, D.; Poole, A.; Fieldhouse, R.J.; Pai, E.F.; Merrill, A.R. Characterization of Vis Toxin, a Novel ADP-Ribosyltransferase from Vibrio splendidus. Biochemistry 2015, 54, 5920–5936.
- Krska, D.; Ravulapalli, R.; Fieldhouse, R.J.; Lugo, M.R.; Merrill, A.R. C3larvin Toxin, an ADP-ribosyltransferase from Paenibacillus larvae. J. Biol. Chem. 2015, 290, 1639–1653.
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47.
- Babii, C.; Mihalache, G.; Bahrin, L.G.; Neagu, A.N.; Gostin, I.; Mihai, C.T.; Sarbu, L.G.; Birsa, L.M.; Stefan, M. A novel synthetic flavonoid with potent antibacterial properties: In vitro activity and proposed mode of action. PLoS ONE 2018, 13, e0194898.
- Cushnie, T.P.T.; Lamb, A.J. Antimicrobial activity of flavonoids. Int. J. Antimicrob. Agents 2005, 26, 343–356.
- Farhadi, F.; Khameneh, B.; Iranshahi, M.; Iranshahy, M. Antibacterial activity of flavonoids and their structure-activity relationship: An update review. Phytother. Res. 2019, 33, 13–40.
- Goel, S.; Khulbe, M.; Aggarwal, A.; Kathuria, A. Recent advances in continuous flow synthesis of heterocycles. Mol. Divers. 2021.
- Egami, H.; Hamashima, Y. Practical and Scalable Organic Reactions with Flow Microwave Apparatus. Chem. Rec. 2019, 19, 157–171.
- Morin, M.A.; Zhang, W.P.; Mallik, D.; Organ, M.G. Sampling and Analysis in Flow: The Keys to Smarter, More Controllable, and Sustainable Fine-Chemical Manufacturing. Angew. Chem. Int. Ed. 2021, 60, 20606–20626.
- Lipinski, C.A. Rule of five in 2015 and beyond: Target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions. Adv. Drug Deliv. Rev. 2016, 101, 34–41.
- Khalifa, S.A.M.; Elshafiey, E.H.; Shetaia, A.A.; El-Wahed, A.A.A.; Algethami, A.F.; Musharraf, S.G.; AlAjmi, M.F.; Zhao, C.; Masry, S.H.D.; Abdel-Daim, M.M.; et al. Overview of Bee Pollination and Its Economic Value for Crop Production. Insects 2021, 12, 688.
- Poppinga, L.; Genersch, E. Molecular pathogenesis of American Foulbrood: How Paenibacillus larvae kills honey bee larvae. Curr. Opin. Insect Sci. 2015, 10, 29–36.
- Applegate, J.R., Jr.; Petritz, O.A. Common and Emerging Infectious Diseases of Honeybees (Apis mellifera). Vet. Clin. Exot. Anim. Pract. 2020, 23, 285–297.
- Ebeling, J.; Knispel, H.; Hertlein, G.; Funfhaus, A.; Genersch, E. Biology of Paenibacillus larvae, a deadly pathogen of honey bee larvae. Appl. Microbiol. Biotechnol. 2016, 100, 7387–7395.
- Genersch, E. American Foulbrood in honeybees and its causative agent, Paenibacillus larvae. J. Invertebr. Pathol. 2010, 103 (Suppl. 1), S10–S19.
- Beims, H.; Bunk, B.; Erler, S.; Mohr, K.I.; Sproer, C.; Pradella, S.; Gunther, G.; Rohde, M.; von der Ohe, W.; Steinert, M. Discovery of Paenibacillus larvae ERIC V: Phenotypic and genomic comparison to genotypes ERIC I–IV reveal different inventories of virulence factors which correlate with epidemiological prevalences of American Foulbrood. Int. J. Med. Microbiol. 2020, 310, 151394.
- Djukic, M.; Brzuszkiewicz, E.; Funfhaus, A.; Voss, J.; Gollnow, K.; Poppinga, L.; Liesegang, H.; Garcia-Gonzalez, E.; Genersch, E.; Daniel, R. How to kill the honey bee larva: Genomic potential and virulence mechanisms of Paenibacillus larvae. PLoS ONE 2014, 9, e90914.
- Funfhaus, A.; Poppinga, L.; Genersch, E. Identification and characterization of two novel toxins expressed by the lethal honey bee pathogen Paenibacillus larvae, the causative agent of American foulbrood. Environ. Microbiol. 2013, 15, 2951–2965.
- Ebeling, J.; Funfhaus, A.; Knispel, H.; Krska, D.; Ravulapalli, R.; Heney, K.A.; Lugo, M.R.; Merrill, A.R.; Genersch, E. Characterization of the toxin Plx2A, a RhoA-targeting ADP-ribosyltransferase produced by the honey bee pathogen Paenibacillus larvae. Environ. Microbiol. 2017, 19, 5100–5116.
- Ebeling, J.; Funfhaus, A.; Genersch, E. The Buzz about ADP-Ribosylation Toxins from Paenibacillus larvae, the Causative Agent of American Foulbrood in Honey Bees. Toxins 2021, 13, 151.
- Turner, M.; Tremblay, O.; Heney, K.A.; Lugo, M.R.; Ebeling, J.; Genersch, E.; Merrill, A.R. Characterization of C3larvinA, a novel RhoA-targeting ADP-ribosyltransferase toxin produced by the honey bee pathogen, Paenibacillus larvae. Biosci. Rep. 2020, 40, BSR20193405.
More
Information
Subjects:
Biochemistry & Molecular Biology; Microbiology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Entry Collection:
Biopharmaceuticals Technology
Revisions:
2 times
(View History)
Update Date:
16 Dec 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No