 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Emilia Caputo | + 3455 word(s) | 3455 | 2021-11-22 08:44:47 | | | |
| 2 | Conner Chen | Meta information modification | 3455 | 2021-12-02 04:52:51 | | | | |
| 3 | Luigi Mandrich | + 3460 word(s) | 6915 | 2021-12-03 15:01:49 | | |
Video Upload Options
Melanoma is a cancer with very poor survival rates, although its treatment has been revolutionized by targeted therapy and immunotherapy. It is a complex disease and here the melanoma complexity has been pointed out as well as the active role of the tumor microenvironment in melanoma progression and its ability to escape to drug treatment. The recent efforts addressed to the development of ex-vivo micro-tissue models able to recapitulate the live conditions of melanoma cells in human patients have been outlined. In particular, the existing ex-vivo melanoma models are reported into the cover picture and include: two-dimensional cell growth in adherent cell culture in a plastic culture dish (a); multicellular melanoma spheroids (b); 3D Skin reconstruct (c); 3D organotypic melanoma spheroids skin model (d); Skin-on-chip (e); ex-vivo tissue slice (f). Further, the use of ex-vivo models as a novel approach for the researcher to investigate the mechanisms underlying tumor biology and immunotherapeutic resistance in metastatic melanoma has been discussed, as well as their high potential for the development of personalized medicine in melanoma treatment.
1. Melanoma
| Gene | Mutated Protein | Frequency (%) * |
Drug/ First Approval Date |
Target | Note |
|---|---|---|---|---|---|
| B-RAF | V600E V600K V600R | ~60 | Vemurafenib/2011 Dabrafenib/2013 Encorafenib/2018 |
BRAFV600E, V600R, V600K kinases | // |
| N-RAS | Q61K Q61R G12D |
~20 | // | // | Tyrosine kinase inhibitors (TKIs) and monoclonal antibodies targeting upstream/downstream NRAS effectors/regulators are in clinical trials |
| MAP2K1/MAP2K2 | E203K E207K | 8 | Trametinib/2013 Cobimetinib/2014 Binimetinib/2017 |
MEK1/MEK2 kinases MEK1 Kinase MEK1/MEK2 kinases |
AZD8330, TAK-733, GDC-0623 are some of MEK1/2 inhibitors in clinical trials |
| PIK3CA | H1047R E545K | ~5 [12] | // | // | class I PI3K, β-sparing PI3K, PI3Kα inhibitors are in clinical trials |
| RAC1 | P29S | ~4 [13] | Under development [14] |
// | Patients carrying RAC1P29S show an increased expression of PD-L1 [15]. Immunotherapy studies by using anti-PD1 or anti PD-L1 antibodies are ongoing |
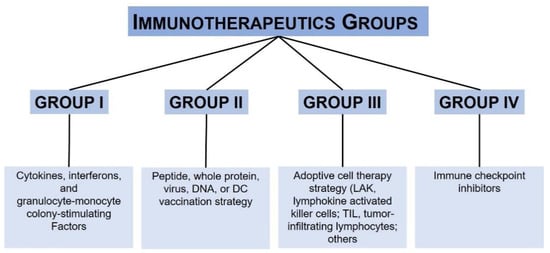
| Immunotherapy | Drug/First Approval Date | Stage |
|---|---|---|
| PD-1 and PD- L1 inhibitor | Nivolumab (Opdivo®)/2014 Pembrolizumab (Keytruda®)/2014 Atezolizumab (Tecentriq®)/2014 |
III |
| CTLA-4 inhibitor | Ipilimumab (Yervoy®)/2011 | III |
| Interferon | Interferon alfa-2b (Intron A®)/2001 Peginterferon alfa-2b (Sylatron®/PEG-Intron®)/2011 |
III |
| Interleukin-2 (IL-2, Proleukin) |
Aldesleukin (Proleukin®)/1998 | III |
| Oncolytic virus | T-VEC (Imlygic®)/2015 | III–IV |
2. Heterogeneity and Plasticity: The Most Striking Melanoma Properties
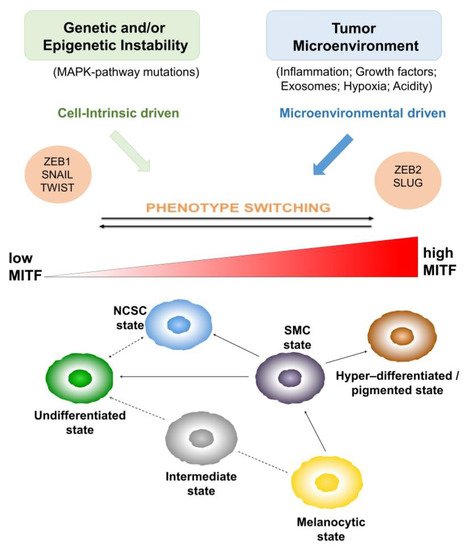
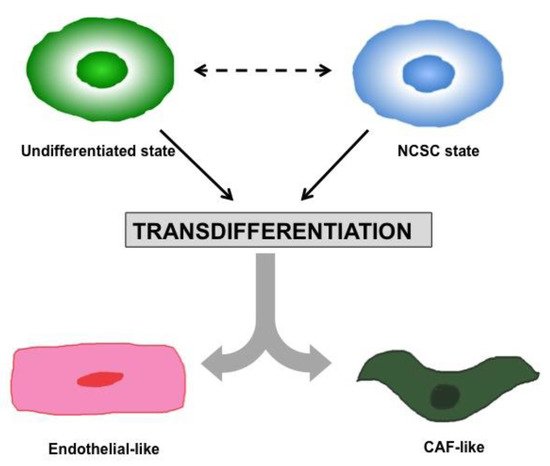
3. Tumor Microenvironment
Tumor microenvironment (TME) plays an active role in melanomagenesis. For instance, overwhelming data on the critical role of the TME in melanoma progression have been reported, supporting the notion that melanoma cells alone are not able to cause disease, but rather need to corrupt and recruit neighboring healthy cell types to use as accessories to their evolution [51-53]. Therefore, the heterogeneous and plastic melanoma cells are only one part of a larger society comprising many other actors, defining the tumor microenvironment as a diversified compartment including neighboring stromal and non-stromal cellular components, the extracellular matrix (ECM) and soluble cues, as schematically illustrated in Figure 3.

Figure 3. Melanoma microenvironment. The main cellular and non-cellular components present in the melanomamicroenvironment are illustrated and indicated in the right part of the figure. The interactions among all of them are reported with an arrow and are described in more detail with a letter indicating: (a) melanoma autocrine growth stimulation and cell survival; (b) melanoma ECM remodeling; (c) stromal reprogramming; (d) regulation of inflammation; (e) leucocyte recruitment; (f) tumor angiogenesis; (g) tumor metabolic reprogramming.
Stroma includes the extracellular matrix (ECM, composed of glycoproteins, proteoglycans, glycosaminoglycans and other macromolecules [54]), growth factors and cytokines, the microvasculature, infiltrating inflammatory cells, and fibroblasts [55]. In melanoma, the stroma appears as desmoplastic (fibroblasts and fibrocytes with mainly fibrillar ECM components accumulation) or myxoid (atypical spindle cells with mainly proteoglycan accumulation [55]) and it is subjected to a constant remodeling by enzymes (collagenases and matrix metalloproteases, MMPs) and by fibroblasts. Interestingly, ECM stiffening, caused by increased collagen (mainly collagen I in the dermis) deposition and crosslinking, has been reported to disrupt the tissue structure, contributing not only to the malignant progression, facilitating tumor dissemination and metastasis, but also to the infiltration of immune cells in tumor sites [56, 57]. The collagen fibers are arranged as vertical fibers in the papillary layer of the dermis, while those in the reticular layer are arranged parallel to the skin surface and are thicker.
Fibroblasts are involved not only in shaping ECM by producing its constituents as collagens and fibrous macromolecules and by degrading them, through releasing proteolytic enzymes, like MMPs; they are also a multifunctional cell type, playing a critical role in maintaining tissue homeostasis and in modulating the immune response. In fact, fibroblasts are involved in the leucocytes recruitment and in the regulation of inflammation, via the secretion of growth factors, cytokines and chemokines [58].
Stroma is activated in cancer, e.g., in wound healing, and fibroblasts inside resemble myofibroblasts observed during wound healing or fibrosis and are called cancer-associated fibroblasts (CAFs) [59, 60].
Interestingly, distinct functional subsets of CAFs, exhibiting tumor-promoting or tumor-suppressing features, have been described, supporting that, as for cancer cells, CAF population is highly heterogeneous [61]. Furthermore, it has been demonstrated that, besides tissue resident fibroblasts, CAFs can also originate from mesenchymal stem cells (MSCs) or stellate cells [62], thus increasing their heterogeneity. MSCs are multipotent progenitor cells originating from the bone marrow. These cells migrate systemically through blood vessels and differentiate into osteoblasts, chondrocytes, or adipocytes. Moreover, they can also differentiate in vascular cells, contributing to angiogenesis in myofibroblasts and, more rarely, in cancer cells themselves. MSCs play a critical role in promoting tissue regeneration and, inside the TME, they modulate the immune response by releasing immunomodulatory cytokines.
The immune cells are another important component of TME. Various immune cell subsets have been identified infiltrating tumors. Among them, tumor-associated macrophages (TAMs) have been identified in all stages of tumor progression, showing antitumoral or pro-tumoral effects according to their inflammatory M1 or immuno-suppressive M2 phenotype, both depending on the microenvironmental stimuli [63, 64].
Eosinophils, as TAMs, are also able to infiltrate tumors and influence tumor progression, inhibiting tumor growth by secreting IL-10 and IL-12, or promoting it by secreting growth factors such as epidermal growth factor (EGF) and transforming growth factor-b1 (TGF-b1) [65]. As tumors grow, myeloid-derived suppressor cells (MDSCs) [66], immunosuppressive precursors of macrophages and dendritic cells (DCs), stimulate the tumor vascularization and unsettle the major mechanisms of immunosurveillance, including tumoral antigen presentation, T cell activation and cytotoxicity.
T cells and natural killer (NK) cells represent the other major subset of tumor infiltrating immune cells. T lymphocytes include three major subtypes: (i) TH lymphocytes divided mainly in two lineages: pro-inflammatory TH1 and anti-inflammatory TH2; (ii) Regulatory T cells (Treg), primarily pro-tumorigenic via their immunosuppressive activity; and (iii) cytotoxic T cells (TC) that kill cancer cells through granzyme and perforin mediated apoptosis [67, 68]. Moreover, a third lineage of effector TH lymphocytes, termed TH17 cells, characterized by their ability to secrete IL-17, have been identified and play a critical role in both anti-tumor immunity and tumorigenesis [69].
T cells are the major players in antitumor immune response. Thanks to the advanced technologies as well as multiplex immunohistochemistry methods [70], and mass cytometry (CyTOF) [71], it has been possible to get a comprehensive phenotyping picture of cells present in human tumor tissues. According to the cancer immunoediting, paradigm T cell infiltration edits the tumor during its progression and tumor evolution depends on the strength and quality of the local immune response at the metastatic site [72]. Intratumoral localization of T cells can be measured as ‘immunoscore’ value, and the high ‘immunoscore’ has been reported to be correlated with improved patient prognosis71. However, T cells can be also found outside the tumor [73, 74], since it has been found that signaling pathways related to tumor cells (intrinsic pathways) or stromal components (extrinsic pathways) could induce T cells to become unable to enter in the tumor bed. This inability, also known as T cell exclusion process, has been indicated as a mechanism of resistance to cancer immunotherapy [75].
Recently, a computational framework has been created on the basis of Tumor Immune Dysfunction and Exclusion (TIDE) to identify factors related to the main mechanisms of tumor immune escape, which could represent a reliable surrogate biomarker to predict the immune checkpoint blockade (ICB) response [76]. Moreover, a signature associated with T cell exclusion and immune evasion has been defined by single-cell RNA sequencing (scRNAseq) of melanoma tumors; it has been demonstrated to be able to predict clinical responses to anti-PD-1 therapy [77].
Furthermore, another important cellular component of TME is represented by the endothelial cells, involved in angiogenesis [78, 79] and vascularization, two important processes involved in cancer cell growth. These cells provide structural integrity to the newly formed vessels and, together with pericytes that ensure their coverage and maturity [80], promote the vascularization inside tumor bed. Endothelial cells not only create the roads for the metastatic dissemination via angiogenesis but they also contribute to chemotherapy resistance through an overexpression of drug efflux pumps thereby decreasing the tumor’s access to the drug [81].
Moreover, cancer-associated adipocytes (CAAs) support cancer growth mainly through secretion of adipokines like adipsin [82] or chemerin [83], as well as proinflammatory cytokines [84] and growth factors. CAAs constitute an important source of lipids for cancer cell membranes and organelles. They are involved in the metabolic reprogramming of cancer cells and in cancer cell invasion, as proteases suppliers [85]. It has been shown that, by producing tumor-promoting cytokines and factors, these cells are able to confer resistance to hormone therapies, chemotherapies, radiotherapies and targeted therapies in breast cancer [86], as well as to contribute to tumor progression of a variety of obesity-associated cancers [87] such as esophagus, gastric, liver, kidney, colorectal, pancreatic, breast, ovarian, prostate, and thyroid cancers. Further, adipocytes, from white adipose tissue, can be recruited to tumor sites where they can differentiate into pericytes and incorporate into vessel walls, thereby contributing to angiogenesis and to tumor proliferation [88].
Additionally, it has been observed that innervated tumors are very aggressive and highly proliferative, with an increased risk of recurrence and metastasis [89]. It is now evident that perineural invasion represents another route for dissemination [90]. Recently, it has been demonstrated that adrenergic nerves promote angiogenesis by activating the angiogenic switch in endothelial cells [91]. Moreover, several studies have described a process inside of a tumor termed axonogenesis, by which cancer cells stimulate the formation of new nerve endings within tumors, through the secretion of neurotrophic factors [92, 93] or by releasing exosomes containing axonal guidance molecules [94]. In return, nerves provide the tumor with neurotransmitters that enhance its growth.
4. Ex Vivo Melanoma Models
4.1. Two-Dimensional (2D) Melanoma Cell Culture
4.2. Three-Dimensional (3D) Melanoma Cell Culture
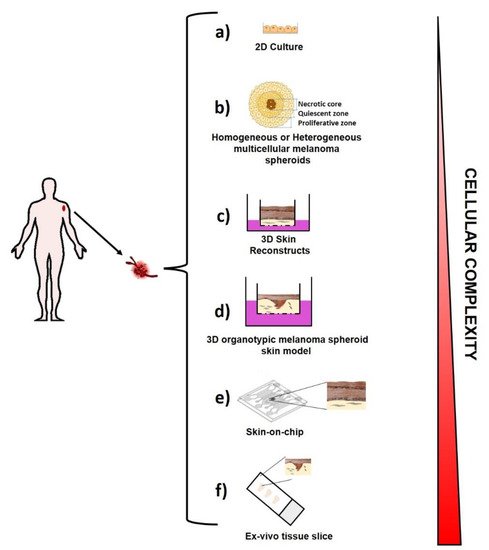
5. Melanoma Immunotherapy and Precision Medicine: Where We Are Today
Although immunotherapy has drastically changed the clinical management of metastatic melanoma, mos patients treated with checkpoint inhibitors (CPIs) do not respond. About 79% of metastatic melanoma patients treated with ipilimumab die within 5 years, while patients treated with the ipilimumab plus nivolumab combined therapy, show a median progression-free survival of 11.5 months compared to 2.9 months observed in patients treated only with ipilimumab [127, 128]. Furthermore, it is not easy to predict which metastatic melanoma patients will be able to respond to with immunotherapy because of the lack of deep understanding of the cellular and molecular mechanisms that lead to PD-1 blockade resistance. Being that this is not the topic of this review, the reader is referred to a recent review on a comprehensive description of the primary and acquired resistance to immune CPIs in metastatic melanoma [129].
Therefore, to move forward with more efficacious personalized treatment and precision medicine, not only predictive markers of response to therapy are under investigation but more pre-clinical models are under development.
Recently, several efforts have been addressed to identify predictive biomarkers of clinical response. Interestingly, gene sequencing studies have discovered markers for monitoring anti-tumor response and therapeutic outcomes after PD-1 blockade failure, like TMB, neoantigen load (NL) or PDL1 expression degree, often associated with an increased response to immunotherapy [130, 131]. Furthermore, a new therapy recently evaluated for melanoma treatment is the oncolytic virus anti-cancer therapy. This therapeutic strategy is based on the ability of oncolytic virus to indirectly lysate tumor cells, leading to the release of soluble antigens and interferons, driving the antitumor immunity. In particular, the attenuated herpes simplex virus-based oncolytic virus talimogene laherparepvec (T-VEC) was FDA approved in 2015, and it is currently used as a local treatment of patients carrying an unresectable advanced stage melanoma [132]. Interestingly, being that oncolytic viruses are able to induce immunogenic tumor cell death, their use in combination with ICIs may represent an interesting and promising strategy for melanoma patients treatment [133].
Similarly, it has been observed that the activation of NF-kB (nuclear factor kappa-light-chain-enhancer of activated B cell) signaling represents a novel potential marker of response to immunotherapy in metastatic melanoma [134]. In particular, it has been observed that a higher mutational load of NFKBIE (NF-kB negative regulator), in codons G34 and G41, only in patients, who were more responsive to anti-PD1 therapy. NFKBIE loss of function culminated in the activation of the NF-kB pathway, which, therefore, can be considered a possible predictive factor of treatment response [135]. Moreover, alterations to DNA damage repair (DDR) pathways have been found associated with a better response to immune checkpoint inhibitors (ICIs).
Furthermore, melanoma heterogeneity is currently treated as another critical parameter of response to immunotherapy. Several studies have shown that patients with less heterogeneous melanoma responded better to the blocking action of anti-CTLA-4 and anti-PD1, supporting the concept that the high heterogeneity implies a major presence of tumor subclones, able to bypass the immune system and thus drug resistance [136].
In addition to the various factors already described, a study conducted on 144 patients with metastatic melanoma included purity and ploidy of the tumor as predictive markers of response to PD1 inhibitors. Specifically, higher tumor purity was associated with tumor progression, while the ploidy was lower in non-responding patients.
The clinical management following immunotherapy failure remains challenging, and a precision medicine (based on specific markers and mutations) is desperately needed in order to create a more personalized treatment [137]. It will help to choose the optimal therapeutic strategies and to predict the consequences of a medical treatment. For instance, the genomic profiling helps to profile the patients and to separate them with the same diagnosis into different groups based on the knowledge of the molecular and cellular mechanisms of the disease [138].
Moreover, moving forward, a precision medicine requires the development of more complex cellular models able to recapitulate more closely the melanoma in human patients [139]; nowadays we are witnessing advances in designing tumor micro-tissues simulating in vivo situations.
6. Melanoma Immunotherapy and Precision Medicine: Where We Are Going in the Tissue Micro-Engineering Era
Advances in the development of melanoma models capable of maintaining the in vivo physiological pressures, where melanoma cells behave as they would in human patients, are offering to the researcher new tools and approaches to better investigate the melanoma biology. Several studies have reported the critical role of TME in modulating T-cell function, particularly in response to PD-1 blockade during melanoma treatment [140] highlighting the necessity of more sophisticated experimental tumor models incorporating key features of the native immune TME, that can be analyzed in real time in order to drive translational research efforts in the clinic.
Recently, human organotypic skin melanoma cultures (OMC) have been developed, by co-culturing decellularized dermis with keratinocytes, fibroblasts and immune cells in the presence of melanoma cells [141]. Interestingly, these human OMCs have been demonstrated to be able to mimic the natural primary human melanoma lesions as well as to be feasible for studying the TME-imprinting mechanisms responsible of melanoma progression. In particular, by using these OMCs, it was demonstrated that the immune cells cDC2s (type 2 conventional dendritic cells) in the TME were melanoma-driven converted into CD14b + DCs.
cDC2s are phenotypically defined as CD1c+CD14− and are able to stimulate cytotoxic T-cell responses [142]. Interestingly, it has been observed that these immune responsive cells were melanoma-induced and converted in CD14+ DCs. These cells are characterized by the expression of genes, such as SSP1, PTGS2 and IL-6, which have been previously associated with immunosuppressive myeloid cells [143, 144], like monocytes and macrophages, having poor T-cell stimulatory ability. Furthermore, the reprogramming of mature cDC2s into CD14+ DCs regulatory macrophage-like cells suggested in this study has been previously proposed only in murine models [145, 146], since the blood cDC2s in human healthy individuals exhibit a low heterogeneity as revealed by single-cell-RNA sequence, and therefore it may not explain the cDC2s phenotypic plasticity observed in these OMCs. Importantly, this study introduced a new tool to use in order to analyze the DCs dynamic interactions with tumor cells inside of the reconstructed TME. This tool could also be used in the future to better define the mechanisms modulating the fate of individual DC subsets within the TME, as well as the migratory nature of DCs towards tumor cells as a stochastic or a chemotactic gradient-driven process induced by the melanoma cells.
Moreover, another study revealed how the organotypic tumor models are powerful tools to evaluate immunotherapy efficacy. In this case, organotypic tumor spheroids grown in collagen hydrogels in a 3-D microfluidic culture system [147] were developed.
In particular, patient- and murine-derived tumor spheroids (MDOTS/PDOTS), retaining tumor-infiltrating lymphoid and myeloid subpopulations, were generated and then used in short-term ex vivo culture to analyze their response to PD-1 blockade treatment, by profiling the secreted cytokines upon ICB treatment [147]. Interestingly, although preliminary data, given the relatively small cohort of samples, a clear relationship between CCL19/CXCL13 cytokines production and immune infiltration was observed. CCL19 has been already reported to be produced from cancer-associated fibroblasts (CAF), while CXCL13 from CD8+ exhausted T cells [40] in melanoma specimens by using single-cell RNA sequencing (RNA-seq). Importantly, this study suggests that both cytokines in PDOTS models may recruit immune- suppressive cells and act as intrinsic resistance mediators to PD-1 blockade.
Actually, CCL19 and CXCL13 cytokines have already been reported to coordinate both humoral and cell-mediated adaptive antitumor immune responses by facilitating the recruitment of naïve T cells and dendritic (CCR7+) and specific B- and T-cell subsets (CXCR5+) to the sites of chronic inflammation [40, 148, 149]. Instead, in this study, these cytokines have been identified as shared acute cytokines to PD1-blockade response, suggesting that future studies need to be performed to highlight the differences between the early and late events of immune response.
Moreover, thanks to the continuous advances in micro-tissue engineering, Votanopoulos et al. were able to generate 3D mixed tumor/node organoids from melanoma patients. The Authors demonstrated that these human experimental models were able to recapitulate the interaction between tumor, host and immune system, representing a feasible platform for personalized immunotherapy screening [150]. In particular, they generated patient-specific immune-enhanced organoids (iPTOs), starting from ten matched melanomas (stage III and IV) and lymph node biospecimens, obtained from the same patient. Further, where it was not possible to obtain lymph nodes from patients, mixed tumor/peripheral T cell organoids were generated starting from the peripheral blood T cell component of the same patient, where tumor was resected.
Additionally, the Authors demonstrated that, when peripheral T cells were circulated through iPTOs and subsequently transferred to naïve PTOs from the same patient, they become able to kill cancer cells, thus suggesting a possible role of iPTOs in generating adaptive immunity.
Although these are preliminary data, given the small numbers of iPTOs examined, they are very promising. It was found a correlation of 85% (6 on 7 patients) between response to immunotherapy observed in the iPTOs and the clinical response of the corresponding patient. Six patients showed melanoma progression while on treatment as their corresponding iPTOs.
All together, these studies demonstrate the enormous potential of ex vivo testing in patient-derived tumor spheroids to identify effective therapeutic combinations to overcome intrinsic resistance to PD-1 blockade. Thus, future adaptations of these models may provide a useful functional approach to drive clinical–translational efforts leading to personalized immunotherapy, lowering its cost, and increasing its effectiveness.
7. Conclusions
It is clear that melanoma is a complex disease, characterized by high heterogeneity and plasticity. Melanoma cells are only a part of a large ecosystem where tumor microenvironment plays an active part on their evolution and on their ability to escape to drug treatment. Therefore, recently several efforts have been addressed to the development of ex-vivo models able to recapitulate the live conditions of melanoma cells in human patients, in order to gain a better understanding of the mechanisms underlying melanoma biology and therapeutic resistance.
Complex 3D-models in microfluidic systems able to mimic the melanoma immune microenvironment have been developed and used as a novel approach by the researcher. Although more complex patient-derived xenografts (PDX) models have been generated [151] to keep the complexity of human tumors, these models are limited by the fact that over time their stromal compartment turns in the murine host one, making more complex the immunotherapy studies [152]. For this reason, in order to investigate on the immune and stromal compartments of the tumor microenvironment, the ex-vivo micro-tissue models are preferred to the PDX ones. Moreover, ex-vivo micro-tissue models have the big advantage to be more efficient and lower in cost than the maintenance of large-scale animal colonies. However, they present limitations in immune cell viability over long-term culture. Thus, more efforts in this field are needed to identify strategies to optimally maintain the various immune populations observed in the tumor microenvironment [153, 154].
Combining ex-vivo micro-tissue models with other techniques, such as single-cell sequencing or advanced microscopy methods, will allow the researcher to highlight our knowledge in immune-tumor cell interactions and immunotherapy and presents the huge potential to pave the way for translational personalized medicine. Therefore, it will be critical to design ex-vivo micro-tissue models compatible with high-throughput molecular analysis such as gene sequencing or mass spectrometry. High-throughput and single-cell gene expression profiling will provide a better understanding of the evolution of tumor cell heterogeneity, as well as of immune landscape dynamicity, when patient-derived organotypic models are cultured within a microfluidic TME.
Finally, in order to use these ex-vivo models in a microfluidic TME as a surrogate for in vivo pre-clinical testing, more studies need to be addressed toward the validation and improved consistency of results. Therefore, it will be critical to repeat studies such as the one from Votanopoulos et al., where the correlation between response to PD-1 blockade in patient-specific immune-enhanced organoids (iPTOs), and in vivo response of the same patient and drug was examined [150]. Patient-derived organotypic models will also likely become a useful tool, particularly for identifying efficacious therapeutic regimes [155]. The establishment of biobanks of patient-derived organoids combined with matched peripheral blood samples for the isolation of circulating immune cells will further support translational research [156-158]. Moreover, these ex-vivo micro-tissue models will be another tool for the researcher, helpful not only to investigate the influence of the microenvironment in tumor progression, but at the same time to allow the researcher to combine, include, or exclude particular TME cell types as well as more straightforward hypothesis testing than using animal models [159].
References
- Carr, S.; Smith, C.; Wernberg, J. Epidemiology and Risk Factors of Melanoma. Surg. Clin. N. Am. 2020, 100, 1–12.
- Chang, A.E.; Karnell, L.H.; Menck, H.R. The National Cancer Data Base Report on Cutaneous and Noncutaneous Melanoma A Summary of 84,836 Cases from the Past Decade. Cancer Interdiscip. Int. J. Am. Cancer Soc. 1998, 83, 1664–1677.
- Mikkelsen, L.H.; Larsen, A.-C.; Von Buchwald, C.; Drzewiecki, K.T.; Prause, J.U.; Heegaard, S. Mucosal malignant melanoma-a clinical, oncological, pathological and genetic. APMIS Surv. 2016, 124, 475–486.
- Rodrigues, M.; De Koning, L.; Coupland, S.E.; Jochemsen, A.G.; Marais, R.; Stern, M.-H.; Valente, A.; Barnhill, R.; Cassoux, N.; Evans, A.; et al. cancers Opinion So Close, yet so Far: Discrepancies between Uveal and Other Melanomas. A Position Paper from UM Cure 2020. Cancers 2019, 11, 1032.
- Elkrief, A.; El Raichani, L.; Richard, C.; Messaoudene, M.; Belkaid, W.; Malo, J.; Belanger, K.; Miller, W.; Jamal, R.; Letarte, N.; et al. Antibiotics are associated with decreased progression-free survival of advanced melanoma patients treated with immune checkpoint inhibitors. Oncoimmunology 2019, 8, e1568812.
- Mazurkiewicz, J.; Simiczyjew, A.; Dratkiewicz, E.; Ziętek, M.; Matkowski, R.; Nowak, D. Stromal cells present in the melanoma niche affect tumor invasiveness and its resistance to therapy. Int. J. Mol. Sci. 2021, 22, 529.
- Roskoski, R. Targeting oncogenic Raf protein-serine/threonine kinases in human cancers. Pharmacol. Res. 2018, 135, 239–258.
- Reddy, B.Y.; Miller, D.M.; Tsao, H. Somatic driver mutations in melanoma. Cancer 2017, 123, 2104–2117.
- Varrone, F.; Caputo, E. The miRNAs role in melanoma and in its resistance to therapy. Int. J. Mol. Sci. 2020, 21, 878.
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134.
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58.
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404.
- Lionarons, D.A.; Hancock, D.C.; Rana, S.; East, P.; Moore, C.; Murillo, M.M.; Carvalho, J.; Spencer-Dene, B.; Herbert, E.; Stamp, G.; et al. RAC1P29S Induces a Mesenchymal Phenotypic Switch via Serum Response Factor to Promote Melanoma Development and Therapy Resistance. Cancer Cell 2019, 36, 68–83.e9.
- Colón-Bolea, P.; García-Gómez, R.; Casar, B. RAC1 Activation as a Potential Therapeutic Option in Metastatic Cutaneous Melanoma. Biomol. 2021, 11, 1554.
- Vu, H.L.; Rosenbaum, S.; Purwin, T.J.; Davies, M.A.; Aplin, A.E. RAC1 P29S regulates PD-L1 expression in melanoma. Pigment Cell Melanoma Res. 2015, 28, 590–598.
- Haanen, J.B.A.G. Immunotherapy of melanoma. Eur. J. Cancer Suppl. 2013, 11, 97–105.
- Choubey, D. Type I interferon (IFN)-inducible Absent in Melanoma 2 proteins in neuroinflammation: Implications for Alzheimer’s disease. J. Neuroinflam. 2019, 16, 236.
- Coventry, B.J. Therapeutic vaccination immunomodulation: Forming the basis of all cancer immunotherapy. Ther. Adv. Vaccines Immunother. 2019, 7, 2515135519862234.
- Sukari, A.; Abdallah, N.; Nagasaka, M. Unleash the power of the mighty T cells-basis of adoptive cellular therapy. Crit. Rev. Oncol. Hematol. 2019, 136, 1–12.
- Chen, C.; Gao, F.-H. Th17 Cells Paradoxical Roles in Melanoma and Potential Application in Immunotherapy. Front. Immunol. 2019, 10, 187.
- Babacan, N.A.; Eroglu, Z. Treatment Options for Advanced Melanoma After Anti-PD-1 Therapy. Curr. Oncol. Rep. 2020, 22, 38.
- Haanen, J.; Ernstoff, M.S.; Wang, Y.; Menzies, A.M.; Puzanov, I.; Grivas, P.; Larkin, J.; Peters, S.; Thompson, J.A.; Obeid, M. Autoimmune diseases and immune-checkpoint inhibitors for cancer therapy: Review of the literature and personalized risk-based prevention strategy. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2020, 31, 724–744.
- Sadozai, H.; Gruber, T.; Hunger, R.E.; Schenk, M. Recent successes and future directions in immunotherapy of cutaneous melanoma. Front. Immunol. 2017, 8, 1–25.
- Grzywa, T.M.; Paskal, W.; Włodarski, P.K. Intratumor and Intertumor Heterogeneity in Melanoma. Transl. Oncol. 2017, 10, 956–975.
- Rambow, F.; Marine, J.C.; Goding, C.R. Melanoma plasticity and phenotypic diversity: Therapeutic barriers and opportunities. Genes Dev. 2019, 33, 1295–1318.
- Vandamme, N.; Berx, G. Melanoma cells revive an embryonic transcriptional network to dictate phenotypic heterogeneity. Front. Oncol. 2014, 4, 1–6.
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28.
- Schatton, T.; Murphy, G.F.; Frank, N.Y.; Yamaura, K.; Gasser, A.M.; Gasser, M.; Zhan, Q.; Jordan, S.; Duncan, L.M.; Weishaupt, C.; et al. Identification of cells initiating human melanomas. Nature 2008, 451, 345–349.
- Büttner, R.; Longshore, J.W.; López-Ríos, F.; Merkelbach-Bruse, S.; Normanno, N.; Rouleau, E.; Penault-Llorca, F. Implementing TMB measurement in clinical practice: Considerations on assay requirements. ESMO Open 2019, 4, 1–12.
- Diener, J.; Sommer, L. Reemergence of neural crest stem cell-like states in melanoma during disease progression and treatment. Stem Cells Transl. Med. 2021, 10, 522–533.
- Quintes, S.; Brinkmann, B.G.; Ebert, M.; Fröb, F.; Kungl, T.; Arlt, F.A.; Tarabykin, V.; Huylebroeck, D.; Meijer, D.; Suter, U.; et al. Zeb2 is essential for Schwann cell differentiation, myelination and nerve repair. Nat. Neurosci. 2016, 19, 1050–1059.
- Denecker, G.; Vandamme, N.; Akay, Ö.; Koludrovic, D.; Taminau, J.; Lemeire, K.; Gheldof, A.; De Craene, B.; Van Gele, M.; Brochez, L.; et al. Identification of a ZEB2-MITF-ZEB1 transcriptional network that controls melanogenesis and melanoma progression. Cell Death Differ. 2014, 21, 1250–1261.
- Koen, E.J.; Collier, A.B. Particle-in-cell simulations of a beam driven plasma. Phys. Plasmas 2012, 4, 1420–1428.
- Lamouille, S.; Xu, J.; Derynck, R. Fakultas Psikologi Dan Sosial Budaya Universitas Islam Indonesia Yogyakarta. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196.
- Caramel, J.; Papadogeorgakis, E.; Hill, L.; Browne, G.J.; Richard, G.; Wierinckx, A.; Saldanha, G.; Sborne, J.; Hutchinson, P.; Tse, G.; et al. A Switch in the Expression of Embryonic EMT-Inducers Drives the Development of Malignant Melanoma. Cancer Cell 2013, 24, 466–480.
- Hao, L.; Ha, J.R.; Kuzel, P.; Garcia, E.; Persad, S. Cadherin switch from E- to N-cadherin in melanoma progression is regulated by the PI3K/PTEN pathway through Twist and Snail. Br. J. Dermatol. 2012, 166, 1184–1197.
- Kim, J.; Lo, L.; Dormand, E.; Anderson, D.J. SOX10 maintains multipotency and inhibits neuronal. Neuron 2003, 38, 17–31.
- Paratore, C.; Goerich, D.E.; Suter, U.; Wegner, M.; Sommer, L. Survival and glial fate acquisition of neural crest cells are regulated by an interplay between the transcription factor Sox10 and extrinsic combinatorial signaling. Development 2001, 128, 3949–3961.
- Civenni, G.; Walter, A.; Kobert, N.; Mihic-Probst, D.; Zipser, M.; Belloni, B.; Seifert, B.; Moch, H.; Dummer, R.; van den Broek, M.; et al. Human CD271-positive melanoma stem cells associated with metastasis establish tumor heterogeneity and long-term growth. Cancer Res. 2011, 71, 3098–3109.
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Ii, M.H.W.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196.
- Graham, D.K.; DeRyckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine-sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785.
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44.
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018, 174, 843–855.e19.
- Müller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.M.; Foppen, M.H.G.; et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014, 5, 5712.
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594.
- Fang, D.; Nguyen, T.K.; Leishear, K.; Finko, R.; Kulp, A.N.; Hotz, S.; Van Belle, P.A.; Xu, X.; Elder, D.E.; Herlyn, M. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005, 65, 9328–9337.
- Madjd, Z.; Erfani, E.; Gheytanchi, E.; Moradi-Lakeh, M.; Shariftabrizi, A.; Asadi-Lari, M. Expression of CD133 cancer stem cell marker in melanoma: A systematic review and meta-analysis. Int. J. Biol. Markers 2016, 31, e118–e125.
- Zabierowski, S.E.; Herlyn, M. Melanoma Stem Cells: The Dark Seed of Melanoma. J. Clin. Oncol. 2008, 26, 2890–2894.
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435.
- Fallahi-Sichani, M.; Becker, V.; Izar, B.; Baker, G.J.; Lin, J.; Boswell, S.A.; Shah, P.; Rotem, A.; Garraway, L.A.; Sorger, P.K. Adaptive resistance of melanoma cells to RAF inhibition via reversible induction of a slowly dividing de-differentiated state. Mol. Syst. Biol. 2017, 13, 905.
- Beaumont, K.; Mohana-Kumaran, N.; Haass, N. Modeling Melanoma In Vitro and In Vivo. Healthcare 2013, 2, 27–46.
- Park, E.S.; Rabinovsky, R.; Carey, M.; Hennessy, B.T.; Agarwal, R.; Liu, W.; Ju, Z.; Deng, W.; Lu, Y.; Woo, H.G.; et al. Integrative analysis of proteomic signatures, mutations, and drug responsiveness in the NCI 60 cancer cell line set. Mol. Cancer Ther. 2010, 9, 257–267.
- Caputo, E.; Maiorana, L.; Vasta, V.; Pezzino, F.M.; Sunkara, S.; Wynne, K.; Elia, G.; Marincola, F.M.; McCubrey, J.A.; Libra, M.; et al. Characterization of human melanoma cell lines and melanocytes by proteome analysis. Cell Cycle 2011, 10, 2924–2936.
- Andrique, L.; Recher, G.; Alessandri, K.; Pujol, N.; Feyeux, M.; Bon, P.; Cognet, L.; Nassoy, P.; Bikfalvi, A. A model of guided cell self-organization for rapid and spontaneous formation of functional vessels. Sci. Adv. 2019, 5, 1–12.
- Ferreira, L.P.; Gaspar, V.M.; Mano, J.F. Design of spherically structured 3D in vitro tumor models -Advances and prospects. Acta Biomater. 2018, 75, 11–34.
- Ramgolam, K.; Lauriol, J.; Lalou, C.; Lauden, L.; Michel, L.; de la Grange, P.; Khatib, A.-M.; Aoudjit, F.; Charron, D.; Alcaide-Loridan, C.; et al. Melanoma spheroids grown under neural crest cell conditions are highly plastic migratory/invasive tumor cells endowed with immunomodulator function. PLoS ONE 2011, 6, e18784.
- Schäfer, M.E.A.; Klicks, J.; Hafner, M.; Rudolf, R. Preparation, Drug Treatment, and Immunohistological Analysis of Tri-Culture Spheroid 3D Melanoma-Like Models. Methods Mol. Biol. 2021, 2265, 173–183.
- Saleh, N.A.; Rode, M.P.; Sierra, J.A.; Silva, A.H.; Miyake, J.A.; Filippin-Monteiro, F.B.; Creczynski-Pasa, T.B. Three-dimensional multicellular cell culture for anti-melanoma drug screening: Focus on tumor microenvironment. Cytotechnology 2021, 73, 35–48.
- Li, Y.; Kumacheva, E. Hydrogel microenvironments for cancer spheroid growth and drug screening. Sci. Adv. 2018, 4, eaas8998.
- Khawar, I.A.; Park, J.K.; Jung, E.S.; Lee, M.A.; Chang, S.; Kuh, H.J. Three Dimensional Mixed-Cell Spheroids Mimic Stroma-Mediated Chemoresistance and Invasive Migration in hepatocellular carcinoma. Neoplasia 2018, 20, 800–812.
- Riffle, S.; Pandey, R.N.; Albert, M.; Hegde, R.S. Linking hypoxia, DNA damage and proliferation in multicellular tumor spheroids. BMC Cancer 2017, 17, 1–12.
- Herter, S.; Morra, L.; Schlenker, R.; Sulcova, J.; Fahrni, L.; Waldhauer, I.; Lehmann, S.; Reisländer, T.; Agarkova, I.; Kelm, J.M.; et al. A novel three-dimensional heterotypic spheroid model for the assessment of the activity of cancer immunotherapy agents. Cancer Immunol. Immunother. 2017, 66, 129–140.
- Li, L.; Fukunaga-Kalabis, M.; Herlyn, M. The three-dimensional human skin reconstruct model: A tool to study normal skin and melanoma progression. J. Vis. Exp. 2011, 12, 1–5.
- Gola, M.; Czajkowski, R.; Bajek, A.; Dura, A.; Drewa, T. Melanocyte stem cells: Biology and current aspects. Med. Sci. Monit. 2012, 18, 155–159.
- Hosaka, C.; Kunisada, M.; Koyanagi-Aoi, M.; Masaki, T.; Takemori, C.; Taniguchi-Ikeda, M.; Aoi, T.; Nishigori, C. Induced pluripotent stem cell-derived melanocyte precursor cells undergoing differentiation into melanocytes. Pigment Cell Melanoma Res. 2019, 32, 623–633.
- Caputo, E.; Miceli, R.; Motti, M.L.; Taté, R.; Fratangelo, F.; Botti, G.; Mozzillo, N.; Carriero, M.V.; Cavalcanti, E.; Palmieri, G.; et al. AurkA inhibitors enhance the effects of B-RAF and MEK inhibitors in melanoma treatment. J. Transl. Med. 2014, 12, 1–9.
- Müller, I.; Kulms, D. A 3D Organotypic Melanoma Spheroid Skin Model. J. Vis. Exp. 2018, 135, e57500.
- Bartfeld, S.; Clevers, H. Organoids as model for infectious diseases: Culture of human and murine stomach organoids and microinjection of helicobacter pylori. J. Vis. Exp. 2015, 2015, 1–9.
- Leslie, J.L.; Young, V.B. A whole new ball game: Stem cell-derived epithelia in the study of host-microbe interactions. Anaerobe 2016, 37, 25–28.
- Garcez, P.P.; Loiola, E.C.; Madeiro da Costa, R.; Higa, L.M.; Trindade, P.; Delvecchio, R.; Nascimento, J.M.; Brindeiro, R.; Tanuri, A.; Rehen, S.K. Zika virus impairs growth in human neurospheres and brain organoids. Science 2016, 352, 816–818.
- Gjorevski, N.; Sachs, N.; Manfrin, A.; Giger, S.; Bragina, M.E.; Ordóñez-Morán, P.; Clevers, H.; Lutolf, M.P. Designer matrices for intestinal stem cell and organoid culture. Nature 2016, 539, 560–564.
- van Duinen, V.; Trietsch, S.J.; Joore, J.; Vulto, P.; Hankemeier, T. Microfluidic 3D cell culture: From tools to tissue models. Curr. Opin. Biotechnol. 2015, 35, 118–126.
- Doherty, E.L.; Aw, W.Y.; Hickey, A.J.; Polacheck, W.J. Microfluidic and Organ-on-a-Chip Approaches to Investigate Cellular and Microenvironmental Contributions to Cardiovascular Function and Pathology. Front. Bioeng. Biotechnol. 2021, 9, 1–14.
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772.
- Valencia, L.; Jorcano, J.L.; Velasco, D. Skin-on-a-chip models: General overview and future perspectives. APL Bioeng. 2021, 5, 030901.
- Guenat, O.T.; Berthiaume, F. Incorporating mechanical strain in organs-on-a-chip: Lung and skin. Biomicrofluidics 2018, 12, 042207.
- Jeffrey, R.; Wozniak; Edward, P.; Riley; Michael, E.; Charness, M.D. Kidney-on-a-chip: Untapped opportunities HHS Public Access. Physiol. Behav. 2019, 176, 139–148.
- Beckwitt, C.H.; Clark, A.M.; Wheeler, S.; Taylor, D.L.; Stolz, D.B.; Griffith, L.; Wells, A. Liver ‘organ on a chip. Exp. Cell Res. 2018, 363, 15–25.
- Meijer, T.G.; Jager, A.; Gent, D.C. Van Ex vivo tumor culture systems for functional drug testing and therapy response prediction-Meijer-2017. Futur. Sci. OA 2017, 3, FSO190.
- Vaira, V.; Fedele, G.; Pyne, S.; Fasoli, E.; Zadra, G.; Bailey, D.; Snyder, E.; Faversani, A.; Coggi, G.; Flavin, R.; et al. Preclinical model of organotypic culture for pharmacodynamic profiling of human tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 8352–8356.
- Bougherara, H.; Mansuet-Lupo, A.; Alifano, M.; Ngô, C.; Damotte, D.; Le Frère-Belda, M.A.; Donnadieu, E.; Peranzoni, E. Real-time imaging of resident T cells in human lung and ovarian carcinomas reveals how different tumor microenvironments control T lymphocyte migration. Front. Immunol. 2015, 6, 1–12.
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guérin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti–PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050.

