Melanoma is the deadliest type of skin cancer. It represents about 5% of all skin tumors and it is the cause of more than 75% of skin cancer deaths worldwide. Patients carrying localized or regional disease show a 5-year relative survival rate value of 98% and 64%, respectively, while this value drastically decreases to 23% in metastatic patients. In order to gain a better knowledge of melanoma evolution, but also to develop successful therapeutic strategies, it is necessary to faithfully recapitulate the in vivo human disease. Therefore, several efforts have been addressed in the development of ex vivo melanoma models able to capture the complex intra-tumor heterogeneity and plasticity in its environmental context.
- melanoma
- tumor microenvironment
- multicellular spheroids
- organotypic melanoma models
- skin-on-chip
1. Melanoma
| Gene | Mutated Protein | Frequency (%) * |
Drug/ First Approval Date |
Target | Note |
|---|---|---|---|---|---|
| B-RAF | V600E V600K V600R | ~60 | Vemurafenib/2011 Dabrafenib/2013 Encorafenib/2018 |
BRAFV600E, V600R, V600K kinases | // |
| N-RAS | Q61K Q61R G12D |
~20 | // | // | Tyrosine kinase inhibitors (TKIs) and monoclonal antibodies targeting upstream/downstream NRAS effectors/regulators are in clinical trials |
| MAP2K1/MAP2K2 | E203K E207K | 8 | Trametinib/2013 Cobimetinib/2014 Binimetinib/2017 |
MEK1/MEK2 kinases MEK1 Kinase MEK1/MEK2 kinases |
AZD8330, TAK-733, GDC-0623 are some of MEK1/2 inhibitors in clinical trials |
| PIK3CA | H1047R E545K | ~5 [12] | // | // | class I PI3K, β-sparing PI3K, PI3Kα inhibitors are in clinical trials |
| RAC1 | P29S | ~4 [13] | Under development [14] |
// | Patients carrying RAC1P29S show an increased expression of PD-L1 [15]. Immunotherapy studies by using anti-PD1 or anti PD-L1 antibodies are ongoing |
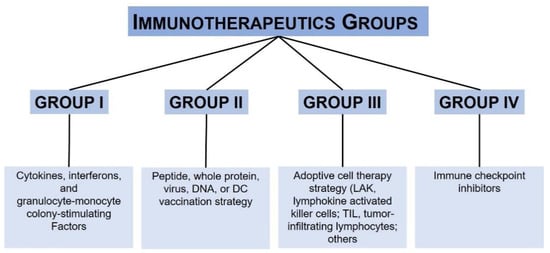
| Immunotherapy | Drug/First Approval Date | Stage |
|---|---|---|
| PD-1 and PD- L1 inhibitor | Nivolumab (Opdivo®)/2014 Pembrolizumab (Keytruda®)/2014 Atezolizumab (Tecentriq®)/2014 |
III |
| CTLA-4 inhibitor | Ipilimumab (Yervoy®)/2011 | III |
| Interferon | Interferon alfa-2b (Intron A®)/2001 Peginterferon alfa-2b (Sylatron®/PEG-Intron®)/2011 |
III |
| Interleukin-2 (IL-2, Proleukin) |
Aldesleukin (Proleukin®)/1998 | III |
| Oncolytic virus | T-VEC (Imlygic®)/2015 | III–IV |
2. Heterogeneity and Plasticity: The Most Striking Melanoma Properties
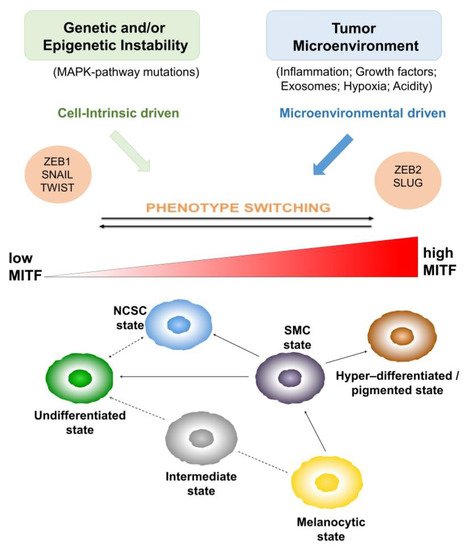
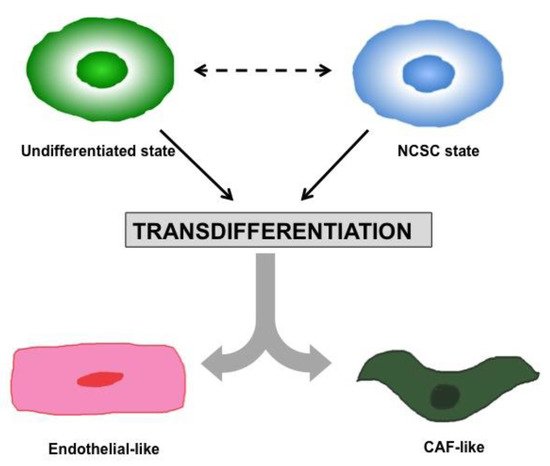
3. Ex Vivo Melanoma Models
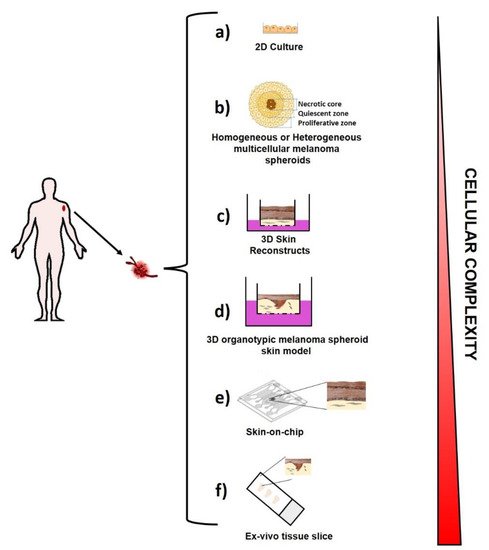
3.1. Two-Dimensional (2D) Melanoma Cell Culture
3.2. Three-Dimensional (3D) Melanoma Cell Culture
 [1]
[1]This entry is adapted from the peer-reviewed paper 10.3390/cancers13225788
References
- Carr, S.; Smith, C.; Wernberg, J. Epidemiology and Risk Factors of Melanoma. Surg. Clin. N. Am. 2020, 100, 1–12.
- Chang, A.E.; Karnell, L.H.; Menck, H.R. The National Cancer Data Base Report on Cutaneous and Noncutaneous Melanoma A Summary of 84,836 Cases from the Past Decade. Cancer Interdiscip. Int. J. Am. Cancer Soc. 1998, 83, 1664–1677.
- Mikkelsen, L.H.; Larsen, A.-C.; Von Buchwald, C.; Drzewiecki, K.T.; Prause, J.U.; Heegaard, S. Mucosal malignant melanoma-a clinical, oncological, pathological and genetic. APMIS Surv. 2016, 124, 475–486.
- Rodrigues, M.; De Koning, L.; Coupland, S.E.; Jochemsen, A.G.; Marais, R.; Stern, M.-H.; Valente, A.; Barnhill, R.; Cassoux, N.; Evans, A.; et al. cancers Opinion So Close, yet so Far: Discrepancies between Uveal and Other Melanomas. A Position Paper from UM Cure 2020. Cancers 2019, 11, 1032.
- Elkrief, A.; El Raichani, L.; Richard, C.; Messaoudene, M.; Belkaid, W.; Malo, J.; Belanger, K.; Miller, W.; Jamal, R.; Letarte, N.; et al. Antibiotics are associated with decreased progression-free survival of advanced melanoma patients treated with immune checkpoint inhibitors. Oncoimmunology 2019, 8, e1568812.
- Mazurkiewicz, J.; Simiczyjew, A.; Dratkiewicz, E.; Ziętek, M.; Matkowski, R.; Nowak, D. Stromal cells present in the melanoma niche affect tumor invasiveness and its resistance to therapy. Int. J. Mol. Sci. 2021, 22, 529.
- Roskoski, R. Targeting oncogenic Raf protein-serine/threonine kinases in human cancers. Pharmacol. Res. 2018, 135, 239–258.
- Reddy, B.Y.; Miller, D.M.; Tsao, H. Somatic driver mutations in melanoma. Cancer 2017, 123, 2104–2117.
- Varrone, F.; Caputo, E. The miRNAs role in melanoma and in its resistance to therapy. Int. J. Mol. Sci. 2020, 21, 878.
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134.
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58.
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404.
- Lionarons, D.A.; Hancock, D.C.; Rana, S.; East, P.; Moore, C.; Murillo, M.M.; Carvalho, J.; Spencer-Dene, B.; Herbert, E.; Stamp, G.; et al. RAC1P29S Induces a Mesenchymal Phenotypic Switch via Serum Response Factor to Promote Melanoma Development and Therapy Resistance. Cancer Cell 2019, 36, 68–83.e9.
- Colón-Bolea, P.; García-Gómez, R.; Casar, B. RAC1 Activation as a Potential Therapeutic Option in Metastatic Cutaneous Melanoma. Biomol. 2021, 11, 1554.
- Vu, H.L.; Rosenbaum, S.; Purwin, T.J.; Davies, M.A.; Aplin, A.E. RAC1 P29S regulates PD-L1 expression in melanoma. Pigment Cell Melanoma Res. 2015, 28, 590–598.
- Haanen, J.B.A.G. Immunotherapy of melanoma. Eur. J. Cancer Suppl. 2013, 11, 97–105.
- Choubey, D. Type I interferon (IFN)-inducible Absent in Melanoma 2 proteins in neuroinflammation: Implications for Alzheimer’s disease. J. Neuroinflam. 2019, 16, 236.
- Coventry, B.J. Therapeutic vaccination immunomodulation: Forming the basis of all cancer immunotherapy. Ther. Adv. Vaccines Immunother. 2019, 7, 2515135519862234.
- Sukari, A.; Abdallah, N.; Nagasaka, M. Unleash the power of the mighty T cells-basis of adoptive cellular therapy. Crit. Rev. Oncol. Hematol. 2019, 136, 1–12.
- Chen, C.; Gao, F.-H. Th17 Cells Paradoxical Roles in Melanoma and Potential Application in Immunotherapy. Front. Immunol. 2019, 10, 187.
- Babacan, N.A.; Eroglu, Z. Treatment Options for Advanced Melanoma After Anti-PD-1 Therapy. Curr. Oncol. Rep. 2020, 22, 38.
- Haanen, J.; Ernstoff, M.S.; Wang, Y.; Menzies, A.M.; Puzanov, I.; Grivas, P.; Larkin, J.; Peters, S.; Thompson, J.A.; Obeid, M. Autoimmune diseases and immune-checkpoint inhibitors for cancer therapy: Review of the literature and personalized risk-based prevention strategy. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2020, 31, 724–744.
- Sadozai, H.; Gruber, T.; Hunger, R.E.; Schenk, M. Recent successes and future directions in immunotherapy of cutaneous melanoma. Front. Immunol. 2017, 8, 1–25.
- Grzywa, T.M.; Paskal, W.; Włodarski, P.K. Intratumor and Intertumor Heterogeneity in Melanoma. Transl. Oncol. 2017, 10, 956–975.
- Rambow, F.; Marine, J.C.; Goding, C.R. Melanoma plasticity and phenotypic diversity: Therapeutic barriers and opportunities. Genes Dev. 2019, 33, 1295–1318.
- Vandamme, N.; Berx, G. Melanoma cells revive an embryonic transcriptional network to dictate phenotypic heterogeneity. Front. Oncol. 2014, 4, 1–6.
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28.
- Schatton, T.; Murphy, G.F.; Frank, N.Y.; Yamaura, K.; Gasser, A.M.; Gasser, M.; Zhan, Q.; Jordan, S.; Duncan, L.M.; Weishaupt, C.; et al. Identification of cells initiating human melanomas. Nature 2008, 451, 345–349.
- Büttner, R.; Longshore, J.W.; López-Ríos, F.; Merkelbach-Bruse, S.; Normanno, N.; Rouleau, E.; Penault-Llorca, F. Implementing TMB measurement in clinical practice: Considerations on assay requirements. ESMO Open 2019, 4, 1–12.
- Diener, J.; Sommer, L. Reemergence of neural crest stem cell-like states in melanoma during disease progression and treatment. Stem Cells Transl. Med. 2021, 10, 522–533.
- Quintes, S.; Brinkmann, B.G.; Ebert, M.; Fröb, F.; Kungl, T.; Arlt, F.A.; Tarabykin, V.; Huylebroeck, D.; Meijer, D.; Suter, U.; et al. Zeb2 is essential for Schwann cell differentiation, myelination and nerve repair. Nat. Neurosci. 2016, 19, 1050–1059.
- Denecker, G.; Vandamme, N.; Akay, Ö.; Koludrovic, D.; Taminau, J.; Lemeire, K.; Gheldof, A.; De Craene, B.; Van Gele, M.; Brochez, L.; et al. Identification of a ZEB2-MITF-ZEB1 transcriptional network that controls melanogenesis and melanoma progression. Cell Death Differ. 2014, 21, 1250–1261.
- Koen, E.J.; Collier, A.B. Particle-in-cell simulations of a beam driven plasma. Phys. Plasmas 2012, 4, 1420–1428.
- Lamouille, S.; Xu, J.; Derynck, R. Fakultas Psikologi Dan Sosial Budaya Universitas Islam Indonesia Yogyakarta. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196.
- Caramel, J.; Papadogeorgakis, E.; Hill, L.; Browne, G.J.; Richard, G.; Wierinckx, A.; Saldanha, G.; Sborne, J.; Hutchinson, P.; Tse, G.; et al. A Switch in the Expression of Embryonic EMT-Inducers Drives the Development of Malignant Melanoma. Cancer Cell 2013, 24, 466–480.
- Hao, L.; Ha, J.R.; Kuzel, P.; Garcia, E.; Persad, S. Cadherin switch from E- to N-cadherin in melanoma progression is regulated by the PI3K/PTEN pathway through Twist and Snail. Br. J. Dermatol. 2012, 166, 1184–1197.
- Kim, J.; Lo, L.; Dormand, E.; Anderson, D.J. SOX10 maintains multipotency and inhibits neuronal. Neuron 2003, 38, 17–31.
- Paratore, C.; Goerich, D.E.; Suter, U.; Wegner, M.; Sommer, L. Survival and glial fate acquisition of neural crest cells are regulated by an interplay between the transcription factor Sox10 and extrinsic combinatorial signaling. Development 2001, 128, 3949–3961.
- Civenni, G.; Walter, A.; Kobert, N.; Mihic-Probst, D.; Zipser, M.; Belloni, B.; Seifert, B.; Moch, H.; Dummer, R.; van den Broek, M.; et al. Human CD271-positive melanoma stem cells associated with metastasis establish tumor heterogeneity and long-term growth. Cancer Res. 2011, 71, 3098–3109.
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Ii, M.H.W.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196.
- Graham, D.K.; DeRyckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine-sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785.
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44.
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018, 174, 843–855.e19.
- Müller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.M.; Foppen, M.H.G.; et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014, 5, 5712.
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594.
- Fang, D.; Nguyen, T.K.; Leishear, K.; Finko, R.; Kulp, A.N.; Hotz, S.; Van Belle, P.A.; Xu, X.; Elder, D.E.; Herlyn, M. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005, 65, 9328–9337.
- Madjd, Z.; Erfani, E.; Gheytanchi, E.; Moradi-Lakeh, M.; Shariftabrizi, A.; Asadi-Lari, M. Expression of CD133 cancer stem cell marker in melanoma: A systematic review and meta-analysis. Int. J. Biol. Markers 2016, 31, e118–e125.
- Zabierowski, S.E.; Herlyn, M. Melanoma Stem Cells: The Dark Seed of Melanoma. J. Clin. Oncol. 2008, 26, 2890–2894.
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435.
- Fallahi-Sichani, M.; Becker, V.; Izar, B.; Baker, G.J.; Lin, J.; Boswell, S.A.; Shah, P.; Rotem, A.; Garraway, L.A.; Sorger, P.K. Adaptive resistance of melanoma cells to RAF inhibition via reversible induction of a slowly dividing de-differentiated state. Mol. Syst. Biol. 2017, 13, 905.
- Beaumont, K.; Mohana-Kumaran, N.; Haass, N. Modeling Melanoma In Vitro and In Vivo. Healthcare 2013, 2, 27–46.
- Park, E.S.; Rabinovsky, R.; Carey, M.; Hennessy, B.T.; Agarwal, R.; Liu, W.; Ju, Z.; Deng, W.; Lu, Y.; Woo, H.G.; et al. Integrative analysis of proteomic signatures, mutations, and drug responsiveness in the NCI 60 cancer cell line set. Mol. Cancer Ther. 2010, 9, 257–267.
- Caputo, E.; Maiorana, L.; Vasta, V.; Pezzino, F.M.; Sunkara, S.; Wynne, K.; Elia, G.; Marincola, F.M.; McCubrey, J.A.; Libra, M.; et al. Characterization of human melanoma cell lines and melanocytes by proteome analysis. Cell Cycle 2011, 10, 2924–2936.
- Andrique, L.; Recher, G.; Alessandri, K.; Pujol, N.; Feyeux, M.; Bon, P.; Cognet, L.; Nassoy, P.; Bikfalvi, A. A model of guided cell self-organization for rapid and spontaneous formation of functional vessels. Sci. Adv. 2019, 5, 1–12.
- Ferreira, L.P.; Gaspar, V.M.; Mano, J.F. Design of spherically structured 3D in vitro tumor models -Advances and prospects. Acta Biomater. 2018, 75, 11–34.
- Ramgolam, K.; Lauriol, J.; Lalou, C.; Lauden, L.; Michel, L.; de la Grange, P.; Khatib, A.-M.; Aoudjit, F.; Charron, D.; Alcaide-Loridan, C.; et al. Melanoma spheroids grown under neural crest cell conditions are highly plastic migratory/invasive tumor cells endowed with immunomodulator function. PLoS ONE 2011, 6, e18784.
- Schäfer, M.E.A.; Klicks, J.; Hafner, M.; Rudolf, R. Preparation, Drug Treatment, and Immunohistological Analysis of Tri-Culture Spheroid 3D Melanoma-Like Models. Methods Mol. Biol. 2021, 2265, 173–183.
- Saleh, N.A.; Rode, M.P.; Sierra, J.A.; Silva, A.H.; Miyake, J.A.; Filippin-Monteiro, F.B.; Creczynski-Pasa, T.B. Three-dimensional multicellular cell culture for anti-melanoma drug screening: Focus on tumor microenvironment. Cytotechnology 2021, 73, 35–48.
- Li, Y.; Kumacheva, E. Hydrogel microenvironments for cancer spheroid growth and drug screening. Sci. Adv. 2018, 4, eaas8998.
- Khawar, I.A.; Park, J.K.; Jung, E.S.; Lee, M.A.; Chang, S.; Kuh, H.J. Three Dimensional Mixed-Cell Spheroids Mimic Stroma-Mediated Chemoresistance and Invasive Migration in hepatocellular carcinoma. Neoplasia 2018, 20, 800–812.
- Riffle, S.; Pandey, R.N.; Albert, M.; Hegde, R.S. Linking hypoxia, DNA damage and proliferation in multicellular tumor spheroids. BMC Cancer 2017, 17, 1–12.
- Herter, S.; Morra, L.; Schlenker, R.; Sulcova, J.; Fahrni, L.; Waldhauer, I.; Lehmann, S.; Reisländer, T.; Agarkova, I.; Kelm, J.M.; et al. A novel three-dimensional heterotypic spheroid model for the assessment of the activity of cancer immunotherapy agents. Cancer Immunol. Immunother. 2017, 66, 129–140.
- Li, L.; Fukunaga-Kalabis, M.; Herlyn, M. The three-dimensional human skin reconstruct model: A tool to study normal skin and melanoma progression. J. Vis. Exp. 2011, 12, 1–5.
- Gola, M.; Czajkowski, R.; Bajek, A.; Dura, A.; Drewa, T. Melanocyte stem cells: Biology and current aspects. Med. Sci. Monit. 2012, 18, 155–159.
- Hosaka, C.; Kunisada, M.; Koyanagi-Aoi, M.; Masaki, T.; Takemori, C.; Taniguchi-Ikeda, M.; Aoi, T.; Nishigori, C. Induced pluripotent stem cell-derived melanocyte precursor cells undergoing differentiation into melanocytes. Pigment Cell Melanoma Res. 2019, 32, 623–633.
- Caputo, E.; Miceli, R.; Motti, M.L.; Taté, R.; Fratangelo, F.; Botti, G.; Mozzillo, N.; Carriero, M.V.; Cavalcanti, E.; Palmieri, G.; et al. AurkA inhibitors enhance the effects of B-RAF and MEK inhibitors in melanoma treatment. J. Transl. Med. 2014, 12, 1–9.
- Müller, I.; Kulms, D. A 3D Organotypic Melanoma Spheroid Skin Model. J. Vis. Exp. 2018, 135, e57500.
- Bartfeld, S.; Clevers, H. Organoids as model for infectious diseases: Culture of human and murine stomach organoids and microinjection of helicobacter pylori. J. Vis. Exp. 2015, 2015, 1–9.
- Leslie, J.L.; Young, V.B. A whole new ball game: Stem cell-derived epithelia in the study of host-microbe interactions. Anaerobe 2016, 37, 25–28.
- Garcez, P.P.; Loiola, E.C.; Madeiro da Costa, R.; Higa, L.M.; Trindade, P.; Delvecchio, R.; Nascimento, J.M.; Brindeiro, R.; Tanuri, A.; Rehen, S.K. Zika virus impairs growth in human neurospheres and brain organoids. Science 2016, 352, 816–818.
- Gjorevski, N.; Sachs, N.; Manfrin, A.; Giger, S.; Bragina, M.E.; Ordóñez-Morán, P.; Clevers, H.; Lutolf, M.P. Designer matrices for intestinal stem cell and organoid culture. Nature 2016, 539, 560–564.
- van Duinen, V.; Trietsch, S.J.; Joore, J.; Vulto, P.; Hankemeier, T. Microfluidic 3D cell culture: From tools to tissue models. Curr. Opin. Biotechnol. 2015, 35, 118–126.
- Doherty, E.L.; Aw, W.Y.; Hickey, A.J.; Polacheck, W.J. Microfluidic and Organ-on-a-Chip Approaches to Investigate Cellular and Microenvironmental Contributions to Cardiovascular Function and Pathology. Front. Bioeng. Biotechnol. 2021, 9, 1–14.
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772.
- Valencia, L.; Jorcano, J.L.; Velasco, D. Skin-on-a-chip models: General overview and future perspectives. APL Bioeng. 2021, 5, 030901.
- Guenat, O.T.; Berthiaume, F. Incorporating mechanical strain in organs-on-a-chip: Lung and skin. Biomicrofluidics 2018, 12, 042207.
- Jeffrey, R.; Wozniak; Edward, P.; Riley; Michael, E.; Charness, M.D. Kidney-on-a-chip: Untapped opportunities HHS Public Access. Physiol. Behav. 2019, 176, 139–148.
- Beckwitt, C.H.; Clark, A.M.; Wheeler, S.; Taylor, D.L.; Stolz, D.B.; Griffith, L.; Wells, A. Liver ‘organ on a chip. Exp. Cell Res. 2018, 363, 15–25.
- Meijer, T.G.; Jager, A.; Gent, D.C. Van Ex vivo tumor culture systems for functional drug testing and therapy response prediction-Meijer-2017. Futur. Sci. OA 2017, 3, FSO190.
- Vaira, V.; Fedele, G.; Pyne, S.; Fasoli, E.; Zadra, G.; Bailey, D.; Snyder, E.; Faversani, A.; Coggi, G.; Flavin, R.; et al. Preclinical model of organotypic culture for pharmacodynamic profiling of human tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 8352–8356.
- Bougherara, H.; Mansuet-Lupo, A.; Alifano, M.; Ngô, C.; Damotte, D.; Le Frère-Belda, M.A.; Donnadieu, E.; Peranzoni, E. Real-time imaging of resident T cells in human lung and ovarian carcinomas reveals how different tumor microenvironments control T lymphocyte migration. Front. Immunol. 2015, 6, 1–12.
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guérin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti–PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050.