Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Conner Chen and Version 1 by Emilia Caputo.
Melanoma is the deadliest type of skin cancer. It represents about 5% of all skin tumors and it is the cause of more than 75% of skin cancer deaths worldwide. Patients carrying localized or regional disease show a 5-year relative survival rate value of 98% and 64%, respectively, while this value drastically decreases to 23% in metastatic patients. In order to gain a better knowledge of melanoma evolution, but also to develop successful therapeutic strategies, it is necessary to faithfully recapitulate the in vivo human disease. Therefore, several efforts have been addressed in the development of ex vivo melanoma models able to capture the complex intra-tumor heterogeneity and plasticity in its environmental context.
- melanoma
- tumor microenvironment
- multicellular spheroids
- organotypic melanoma models
- skin-on-chip
1. Melanoma
Melanoma is the deadliest type of skin cancer. It represents about 5% of all skin tumors and it is the cause of more than 75% of skin cancer deaths worldwide. Patients carrying localized or regional disease show a 5-year relative survival rate value of 98% and 64%, respectively, while this value drastically decreases to 23% in metastatic patients [1].
Melanomas originate from the malignant transformation of the melanocytes and they are mainly classified in three subtypes according to the localization of melanocytes undergoing the transformation: 1. cutaneous melanoma (CM), from skin melanocytes; 2. uveal melanoma (UM), from melanocytes in the choroid, ciliary body, and iris of the eye, and 3. mucosal melanomas (MM) from melanocytes in mucosal membranes [2,3,4][2][3][4]. CM represents 91.2%, while UM 5.3% and MM 1.3% of all melanomas recorded in the USA. Due to its prevalence, CM (hereafter melanoma) is the most studied subtype among the three and it will be the focus of this review.
Melanoma is a complex disease consisting of a multistep process, involving the accumulation of genetic and/or epigenetic somatic modifications and exposition to environmental factors, where not only melanoma cells themselves, but constant interactions occurring between tumor cells and their surroundings play a crucial role in disease dissemination, therapy resistance, and mortality. Indeed, it is a dynamic process where inter- and intra-tumoral heterogeneity, phenotypic plasticity, stromal reprogramming, and microbiome [5] are among some of the key drivers of melanoma progression. Over the last decade, remarkable advances have been made in expounding melanoma etiology and pathogenesis, promoting the identification and validation of novel drug targets and biomarkers [6]. Today, surgery remains the gold standard treatment for the primary melanoma, mainly for thin melanomas, with low risk of dissemination and long-term survival benefits. On the other hand, metastatic melanoma therapy has been revolutionized by developments in targeted therapy and immunotherapy.
Targeted therapy approaches have been possible thanks to the identification of important somatic mutations, referred to as ‘driver mutations’, conferring advantages to melanoma growth and progression. The most studied and well-characterized driver mutations in melanoma have been found mainly in genes involved in the Mitogen-Activated Protein Kinase (MAPK) [7,8,9][7][8][9] and in the Phosphoinositide 3-Kinase/protein Kinase B (PI3K/AKT) pathways [10,11][10][11]. Among these genes, B-RAF, encoding the proto-oncogene serine/threonine kinase, has been the first well-studied driver mutation to be used in targeted therapy approach for melanoma treatment and, over time, different drugs have been developed against B-RAF mutated proteins, from vemurafenib to dabrafenib and the recent encorafenib. The most significant somatic driver mutations and the corresponding designed targeting drugs are reported in Table 1.
Table 1. Most significant somatic driver mutations and drugs. In the columns were indicated the gene names, the corresponding mutated proteins and the percentage of the melanoma patients carrying the indicated mutations as well as drugs targeting the mutated proteins, drug approval date, drug targets and some notes.
| Gene | Mutated Protein | Frequency (%) * |
Drug/ First Approval Date |
Target | Note |
|---|---|---|---|---|---|
| B-RAF | V600E V600K V600R | ~60 | Vemurafenib/2011 Dabrafenib/2013 Encorafenib/2018 |
BRAFV600E, V600R, V600K kinases | // |
| N-RAS | Q61K Q61R G12D |
~20 | // | // | Tyrosine kinase inhibitors (TKIs) and monoclonal antibodies targeting upstream/downstream NRAS effectors/regulators are in clinical trials |
| MAP2K1/MAP2K2 | E203K E207K | 8 | Trametinib/2013 Cobimetinib/2014 Binimetinib/2017 |
MEK1/MEK2 kinases MEK1 Kinase MEK1/MEK2 kinases |
AZD8330, TAK-733, GDC-0623 are some of MEK1/2 inhibitors in clinical trials |
| PIK3CA | H1047R E545K | ~5 [12] | // | // | class I PI3K, β-sparing PI3K, PI3Kα inhibitors are in clinical trials |
| RAC1 | P29S | ~4 [13] | Under development [14] |
// | Patients carrying RAC1P29S show an increased expression of PD-L1 [15]. Immunotherapy studies by using anti-PD1 or anti PD-L1 antibodies are ongoing |
* Frequency from the TCGA melanoma cohort.
Immunotherapy is another efficient treatment option for metastatic melanoma patients, because of the high immunogenicity of this tumor. Four main groups of immunotherapeutic treatments are currently available for melanoma treatment [16], as illustrated in Scheme 1.
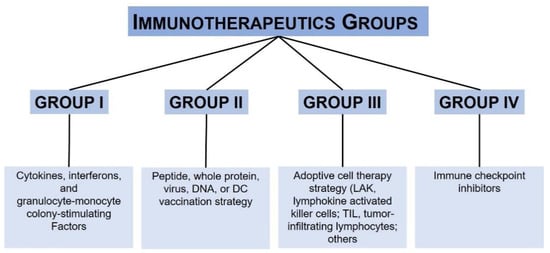
In particular, antibodies directed to specific immune checkpoints such as anti-programmed cell death 1 (PD-1) and anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) brought a statistically significant benefit in terms of overall survival (OS), progression-free survival (PFS) and overall response rate (ORR) compared to chemotherapy.
Both target based therapeutics and the immune checkpoint inhibitors [23] have drastically changed the clinical management of melanoma and improved melanoma patient outcome, since their FDA approvals. The current immunotherapeutic drugs for melanoma treatment are summarized in Table 2.
Table 2. Approved immunotherapy strategies for melanoma. In the columns were indicated the immunotherapy strategy, the drug with the approval date and the melanoma stage.
| Immunotherapy | Drug/First Approval Date | Stage |
|---|---|---|
| PD-1 and PD- L1 inhibitor | Nivolumab (Opdivo®)/2014 Pembrolizumab (Keytruda®)/2014 Atezolizumab (Tecentriq®)/2014 |
III |
| CTLA-4 inhibitor | Ipilimumab (Yervoy®)/2011 | III |
| Interferon | Interferon alfa-2b (Intron A®)/2001 Peginterferon alfa-2b (Sylatron®/PEG-Intron®)/2011 |
III |
| Interleukin-2 (IL-2, Proleukin) |
Aldesleukin (Proleukin®)/1998 | III |
| Oncolytic virus | T-VEC (Imlygic®)/2015 | III–IV |
However, they left a big challenge for investigators and clinicians in overcoming the drug resistance and disease recurrence issues responsible of the therapeutic plateau we are in today.
Recent advances in tumor micro-tissue engineering are providing novel insights in the melanoma biology and in its complexity, which can be translated in the development of innovative and successful target-based and immune therapies.
2. Heterogeneity and Plasticity: The Most Striking Melanoma Properties
Melanoma is characterized by a high heterogeneity [24] and plasticity [25]. The intra-tumor heterogeneity is the product of the high irreversible genetic instability of melanoma cells as well as of their ability to undergo reversible phenotypic changes. The high genetic instability [8] generates the necessary genetic modification leading to the irreversible cell-intrinsic phenotype switching ability of melanoma cells, while on the other hand, their reversible ability to switch phenotype [26] is driven by micro-environmental cues. Both, cell-intrinsic and microenvironmental-driven, are more generally referred to as ‘phenotype switching’, which is, actually, the better model explaining the dynamic melanoma evolution [26], as explained below. It includes both the clonal evolution [27] and the cancer-stem cells [28] models.
Further, melanoma cells display an extreme plasticity. They are able to activate a plastic network of signal transduction pathways passing from one path to another one, in order to keep the continuous transmission of survival signals even in hostile environments. In addition, their ability to transdifferentiate to a variety of states under different circumstances represents a further mechanism underlying their incredible plasticity, as explained in the following sections.
Nevertheless, melanoma cells are able to influence the tumor microenvironment by a stromal reprogramming mechanism, which is responsible for their long-term growth and drug resistance.
Melanoma evolution and phenotype switching. Melanoma cells derive from the melanocytes transformation, which is accompanied by the accumulation of a high number of driver mutations, as demonstrated by cancer genome deep sequencing [29]. This genetic instability [8] shapes the necessary genetic alteration driving to the irreversible cell-intrinsic phenotype switching ability of melanoma cells responsible for melanoma progression.
Moreover, it has been demonstrated that melanoma progression is associated with the reactivation of melanocyte differentiation program [30]. Briefly, melanocytes are the product of an active epithelial to mesenchymal transition (EMT) program occurring during embryogenesis, in the (neuroepithelial) neural crest stem cells (NCSCs), despite the fact that melanocytes are not true epithelial cells. The NCSCs are a multipotent, migratory, and transient cell population. During embryonic development, these cells migrate through the vertebrate embryo and infiltrate different organs where they are capable to differentiate in various cell lineages including melanocytes [26]. It has been demonstrated that ZEB2, one among the EMT-inducing transcription factors (EMT-TFs), is essential for terminal differentiation in vivo of NCSCs in melanocytes, Schwann cells and oligodendrocytes, through the upregulation of microphthalmia-associated transcription factor, MITF [31,32][31][32].
EMT is a cellular program crucial not only during embryogenesis, but also in the course of fibrosis, wound healing and carcinogenesis [26,33][26][33]. It induces a down-regulation of epithelial markers and an upregulation of mesenchymal markers in the cells, accompanied by a morphological change from an epithelioid towards a mesenchymal/spindle cell shape as well as by a remodeling of cell-cell and cell-matrix interactions with subsequent enhanced cell motility and migration [34].
The molecular pathways activated by EMT-TFs and MITF, during the melanocyte differentiation program, may be reactivated during melanomagenesis, which explains melanoma heterogeneity and plasticity. It has been found that likely in NC-derived melanoblasts, the switch from E to N-Cadherin does occur in a subset of melanomas and it is induced by ZEB1, TWIST and SNAIL EMT-TFs [26,35,36][26][35][36]. These findings support that melanoma progression is not founded only on irreversible clonal or lineage-driven remodeling, but can be induced by reversible and functional reprograming of signaling pathways, activated by EMT-TFs and MITF, according to the ‘phenotype switching’ model as showed in Figure 1.
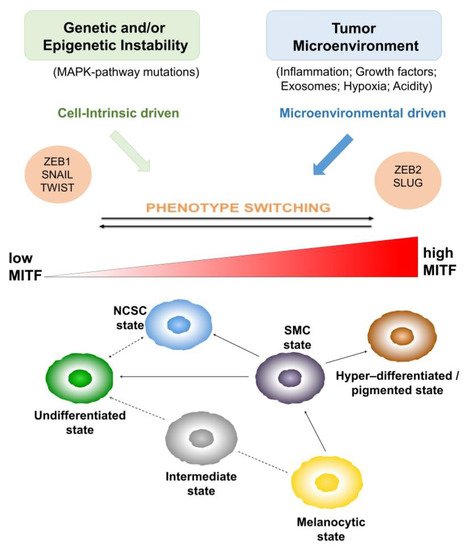
Figure 1. Melanoma evolution and phenotype switching. Melanoma heterogeneity as the product of cell-intrinsic and microenvironmental driven ‘phenotype switching’. The role of epithelial-to-mesenchymal transition-inducing transcription factors (EMT-TFs) and of microphthalmia-associated transcription factor (MITF) is highlighted in the ‘phenotype switching’ model explaining melanoma evolution. The revised MITF rheostat model, including six different phenotypic states, explaining the oscillation between differentiated vs invasive melanoma cells through the reactivation of melanocyte differentiation program in melanoma is illustrated on the bottom.
Moreover, it has been observed that CD271 and SOX10, two factors [37,38][37][38] associated with NCSC regulatory networks [30], are highly expressed in human melanoma and their expression is correlated with a high metastatic potential and a worse patient prognosis [39].
Despite the fact that melanoma phenotypic diversity and plasticity have been known for many years, the molecular characterization of the different phenotypic states has been pointed out with the cloning of MITF genes and with the discovery of its role in the reversible phenotype switching of melanoma cells between an MITF-positive/drug sensitive ‘proliferative’ state and an MITF-low/drug resilient ‘invasive’ cell state. Moreover, it has been observed that MITF-depleted cells showed a more stem cell-like phenotype, an increased plasticity and a reduced proliferation, promoting tumor progression, while cells expressing high levels of MITF stimulated proliferation and differentiation [34], as illustrated in Figure 1.
Single-cell RNA sequencing has further contributed to the identification of the transcriptional programs underlying melanoma phenotypic heterogeneity [40]. These studies confirmed the key role of MITF for distinct phenotypic states, in particular, its correlation to a differentiation gene-expression program. On the other hand, these studies highlighted the opposite role of AXL, a receptor tyrosine kinase belonging to TAM family in promoting the invasive, dedifferentiated drug resistant phenotype [41,42,43,44][41][42][43][44].
Interestingly, it has been observed that melanomas classified as MITFHigh contained a small amount of cells expressing AXLHigh/MITFLow program. This small population increased upon treatment with BRAF inhibitors (BRAFi) as single agents or in combination with MEK inhibitors (MEKi), along with a distinct resistant population of MET-high cells. Moreover, the AXLHigh/MITFLow cell population was associated with increased numbers of cancer associated fibroblasts (CAFs), while MITFHigh melanomas showed a reduced CAF infiltration.
Actually, the phenotypic states of melanoma cells are not limited to an MITF-positive/drug sensitive ‘proliferative’ and an MITF-low/drug resilient ‘invasive’ cell state; however, to date, a revised MITF rheostat model, including six different phenotypic states, has been reported.
As shown in Figure 1, the states are ranked in relation to MITF and SOX10 expression. Starting from the MITFLow/SOX10Low undifferentiated state, the most undifferentiated melanoma cells, having lost expression of both melanocytic transcription factors, SOX10 and MITF, to the MITFLow/SOX10Medium neural crest stem cell (NCSC) state, to the MITFMedium/SOX10Medium starved melanoma cell (SMC), intermediate and melanocytic states to the MITFHigh/SOX10Low hyper-differentiated state. This classification, according to the MITF expression or differentiation, does not rule out the potential of each plastic state to generate another one. Indeed, it has been demonstrated that hyper-differentiated, NCSC and undifferentiated states [43] can be generated starting from an SMC state, as a precursor.
Lineage plasticity and transdifferentiation. Melanoma cells display an incredible plasticity. Different subsets of melanoma cells exhibiting a dedifferentiated state and stem-like properties, akin to their NCSC precursors, have been identified [45]. In particular, it has been demonstrated that these stem-like subpopulations display NCSC molecular features (i.e., KDM5B [45], CD133 [46[46][47],47], CD20 [46[46][48],48], NGFR [49,50][49][50] and AQP1), as well as biological properties such as high plasticity, migratory capacity, invasiveness and a general loss of pigmentation. Along with the lineage plasticity, transdifferentiation is another process described in melanoma cells, underlying their striking plasticity. Melanoma cells are able to transdifferentiate by exiting the melanocytic lineage to a different cell lineage like endothelial cells or CAFs [26,40][26][40], as illustrated in Figure 2.
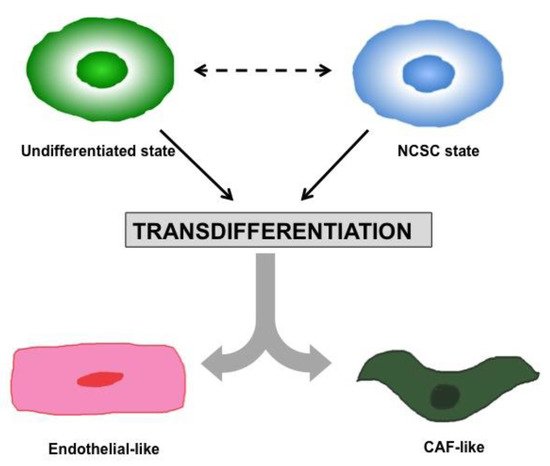
Figure 2. Melanoma Plasticity and Transdifferentiation. Melanoma cells are able to transdifferentiate by exiting the melanocytic lineage to a different cell lineage like endothelial cells or CAFs.
Therefore, these subpopulations are able to adopt, transiently or permanently, different cellular states, each with implications on the proliferative abnormality, stromal reprogramming, angiogenesis, tumor sustaining inflammation and drug sensitivity, facilitating melanoma growth and progression. For instance, subpopulations of melanoma cells expressing high levels of EGFR and NGFR have been identified inside of tumors before therapy; it has been demonstrated that they are responsible for therapeutic relapse [49].
Moreover, melanoma cells are able to secrete growth factors and cytokines normally produced by stromal fibroblasts, macrophages, neutrophils and monocytes, promoting cell survival in an autocrine manner and influencing the tumor microenvironment in a paracrine loop. This mechanism, adopted by melanoma cells, is also known as stromal reprogramming.
3. Ex Vivo Melanoma Models
3.1. Two-Dimensional (2D) Melanoma Cell Culture
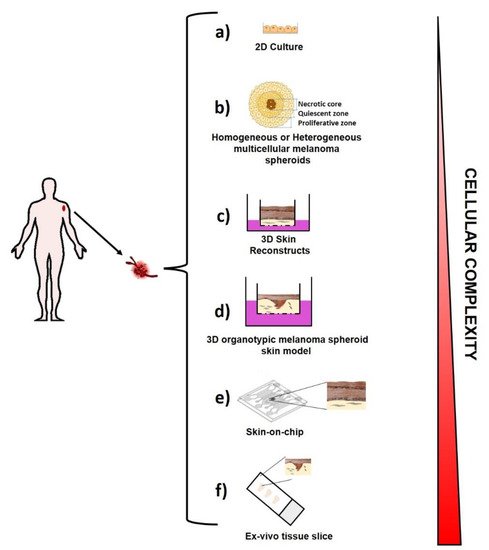
For several years, melanoma cells by themselves have been studied and largely characterized by using “traditional” melanoma cell lines established from patients and cultured on plastic (Figure 4a), with high levels of oxygen and nutrients.
Although this cell system does not provide information on cell-cell and cell–extracellular matrix interactions and on the tumor complexity, as well as on the melanoma behavior in vivo, this approach showed great utility in translational melanoma research. Comparative ‘omic’ studies aimed at characterizing melanoma cells established from melanoma patients at different clinical stages vs. melanocytes, and/or pre-malignant nevus cells able to identify biomarkers associated to the different cell lines examined [95,96,97][51][52][53].
Up to date, more than 2000 melanoma cell lines have been generated. Since these monocultures are free from other contaminating cells, the extensive genetic and genomic analysis that has been performed for the most of them, provided a comprehensive landscape of genes and pathways associated to melanoma progression and its drug resistance ability. In addition, the possibility to grow these cells in co-culture conditions, by using trans-well plates, allowed gaining a better understanding of the melanoma cell behavior upon a given insult.
Although there are several limits of the traditional 2D cell culture, such as the lack of heterogeneity, the different behavior of melanoma cells grown on plastic compared to the one observed in vivo, the high genetic variability with consequent lack of reproducibility of the experimental results depending on the genetic drift, occurring in long-term passaged cell lines, this approach remains very useful for the initial high-throughput screening to identify potential hits worth of further examination.
3.2. Three-Dimensional (3D) Melanoma Cell Culture
Multicellular Spheroids. Multicellular Spheroids (MCSs) consist of 3D cellular aggregates of homogeneous or heterogeneous cell populations derived from tissue fragments mechanically and/or enzymatically partially digested, as illustrated in Figure 4b. MCSs are obtained in the absence of a scaffolding material, as cultured cells produce their own ECM and can be used to generate either homogeneous tumor models by starting from solely cancer cells cultures, or more sophisticated heterotypic spheroids by starting from cancer cells cultures with components of the TME like fibroblasts, endothelial cells [98][54] or immune cells. Different techniques have been developed to generate these models in laboratory, such as the forced floating methods in non-adherent plates, the hanging drop method, the use of scaffolds and matrices, or even more sophisticated methods using microfluidic systems [99][55].
Current three-dimensional melanoma models are composed of melanoma cells only (melanoma spheroids) [100][56] or they are more sophisticated, including multiple cell types to reproduce human skin equivalents with skin-like organization. An intermediate spheroid-based model has also been developed, consisting of tri-cultures of human fibroblasts, keratinocytes, and melanoma cells. These systems have the advantage of being reliably reproduced without the need of special equipment, since they are made of cell lines and therefore can be generated by using standard culture procedures. Moreover, this kind of model can be helpful to investigate the different aspects of skin and early melanoma formation as to test drug compounds [101][57]. Further, melanoma multicellular spheroids model composed of melanoma cells, fibroblasts, and macrophages have been generated by liquid-overlay technique using agarose gel. These models have been helpful in the examination of stromal cell influence on their size, growth, viability and morphology, compared to the melanoma monocellular spheroids [102][58].
Recently, several efforts are addressed to developing biomimetic hydrogel scaffolds that can then be used to encapsulate the MCSs in order to provide a more complex tumor model, where the biophysical and biochemical cues are enclosed, simulating the behavior of the ECM, that it is known to play an important role in the modulation of cancer cell behavior [103][59].
Although these models recapitulate the TME heterogeneity [104][60], oxygen gradients [105][61], and immune infiltration [106][62], they are limited by the lack of control over the 3D culture environment, being that they are generated by the self-assembling of cells.
3D skin reconstructs. This 3D model captures the melanoma heterogeneity and the complex intra-cellular interactions similar to the one occurring in in vivo human disease. It includes an “epidermis” containing stratified, differentiated keratinocytes, a functional basement membrane, and a “dermis” with fibroblasts embedded in collagen I, the most prevalent extracellular matrix (ECM) present in the human skin [107][63] (Figure 4c). However, in order to generate 3D skin reconstructs in the laboratory, the ability to obtain melanoma cells, keratinocytes, fibroblasts and melanocytes in viable culture is critical. Fibroblasts and melanocytes can also be derived from human skin, but can either come from embryonic stem cells (ESCs) [108][64] or induced pluripotent stem (iPS) cells [109][65]. The 3D skin reconstruct models are helpful tools for invasion and metastasis studies as well as for analysis of drug effects on melanoma cells [110][66].
Organotypic Melanoma Models Like Organoids. 3D organotypic melanoma models have been developed [111][67] to reproduce ex vivo the complexity of melanoma (Figure 4d). They are considered the more physiological 3D culture models. Similar to tissue like organoids [112[68][69][70],113,114], they represent an innovative approach for melanoma modeling studies and anticancer drug testing. Several efforts are ongoing in order to develop novel synthetic analogous ECM, controllable for allowing a fine tuning of matrix constituents [115][71]. These approaches permit one to mimic the organ topography, the cancer cells’ mechanical forces, the stiffness, functionality, and complexity of matrix much better than 2D or even 3D culture systems [116][72].
Skin-on-chip. Since organotypic melanoma models lack parameters such as fluid shear stress and hydrostatic pressure, which are able to greatly influence cell behavior in the physiological conditions, several efforts have been addressed into the development of microfluidic systems [117][73]. These cell culture systems, also known as organ-on-a-chip, are made of hollow microchannels populated by living cells and continuously perfused [118][74]. To date, skin-on-chip [119][75] have been successfully modeled in microfluidic devices (Figure 4e), as well as lung alveoli [120][76], human kidney tubules [121][77], and liver [122][78]. These systems show the big advantage to reproduce a spatio-temporally controlled microenvironment, where all the molecular, biophysical and cellular components can be tuned according to the physiologically relevant parameters Furthermore, they represent a feasible tool for drug efficiency and toxicity assessment.
Ex vivo tissue slices [123][79]. They represent a further tool, by which the tissue 3D architecture and pathway activity is preserved although for short time [124][80] (Figure 4f). This tool has been revealed to feasibly track T cells and identifying the extracellular matrix as the major stromal component influencing T cell migration in fresh human tumor tissues [125][81]. Furthermore, the analysis of ex vivo tissue slices by dynamic imaging microscopy allowed us to highlight the mechanism underlying T cell exclusion by examining the interaction between endogenous CD8 T cells and tumor-associated macrophages (TAMs) inside the tumor stroma. These studies translated in a murine model showed that TAMs depletion enhanced the efficacy of anti–PD-1 immunotherapy [126][82]. Thus, this tool may be used in studies of screening for novel immunotherapy agents and in the T cells monitoring inside of the tumor.
 [1]
[1]
[1]Figure 4. Ex-vivo Melanoma Models. A schematic representation of the existing ex-vivo melanoma models: two-dimensional cell growth in adherent cell culture in a plastic culture dish (a); multicellular melanoma spheroids (b); 3D Skin reconstruct (c); 3D organotypic melanoma spheroids skin model (d); Skin-on-chip (e); Ex-vivo tissue slice (f).
References
- Francesca Varrone; Luigi Mandrich; Emilia Caputo; Melanoma Immunotherapy and Precision Medicine in the Era of Tumor Micro-Tissue Engineering: Where Are We Now and Where Are We Going?. Cancers 2021, 13, 5788, 10.3390/cancers13225788.Carr, S.; Smith, C.; Wernberg, J. Epidemiology and Risk Factors of Melanoma. Surg. Clin. N. Am. 2020, 100, 1–12.
- Chang, A.E.; Karnell, L.H.; Menck, H.R. The National Cancer Data Base Report on Cutaneous and Noncutaneous Melanoma A Summary of 84,836 Cases from the Past Decade. Cancer Interdiscip. Int. J. Am. Cancer Soc. 1998, 83, 1664–1677.
- Mikkelsen, L.H.; Larsen, A.-C.; Von Buchwald, C.; Drzewiecki, K.T.; Prause, J.U.; Heegaard, S. Mucosal malignant melanoma-a clinical, oncological, pathological and genetic. APMIS Surv. 2016, 124, 475–486.
- Rodrigues, M.; De Koning, L.; Coupland, S.E.; Jochemsen, A.G.; Marais, R.; Stern, M.-H.; Valente, A.; Barnhill, R.; Cassoux, N.; Evans, A.; et al. cancers Opinion So Close, yet so Far: Discrepancies between Uveal and Other Melanomas. A Position Paper from UM Cure 2020. Cancers 2019, 11, 1032.
- Elkrief, A.; El Raichani, L.; Richard, C.; Messaoudene, M.; Belkaid, W.; Malo, J.; Belanger, K.; Miller, W.; Jamal, R.; Letarte, N.; et al. Antibiotics are associated with decreased progression-free survival of advanced melanoma patients treated with immune checkpoint inhibitors. Oncoimmunology 2019, 8, e1568812.
- Mazurkiewicz, J.; Simiczyjew, A.; Dratkiewicz, E.; Ziętek, M.; Matkowski, R.; Nowak, D. Stromal cells present in the melanoma niche affect tumor invasiveness and its resistance to therapy. Int. J. Mol. Sci. 2021, 22, 529.
- Roskoski, R. Targeting oncogenic Raf protein-serine/threonine kinases in human cancers. Pharmacol. Res. 2018, 135, 239–258.
- Reddy, B.Y.; Miller, D.M.; Tsao, H. Somatic driver mutations in melanoma. Cancer 2017, 123, 2104–2117.
- Varrone, F.; Caputo, E. The miRNAs role in melanoma and in its resistance to therapy. Int. J. Mol. Sci. 2020, 21, 878.
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134.
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58.
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404.
- Lionarons, D.A.; Hancock, D.C.; Rana, S.; East, P.; Moore, C.; Murillo, M.M.; Carvalho, J.; Spencer-Dene, B.; Herbert, E.; Stamp, G.; et al. RAC1P29S Induces a Mesenchymal Phenotypic Switch via Serum Response Factor to Promote Melanoma Development and Therapy Resistance. Cancer Cell 2019, 36, 68–83.e9.
- Colón-Bolea, P.; García-Gómez, R.; Casar, B. RAC1 Activation as a Potential Therapeutic Option in Metastatic Cutaneous Melanoma. Biomol. 2021, 11, 1554.
- Vu, H.L.; Rosenbaum, S.; Purwin, T.J.; Davies, M.A.; Aplin, A.E. RAC1 P29S regulates PD-L1 expression in melanoma. Pigment Cell Melanoma Res. 2015, 28, 590–598.
- Haanen, J.B.A.G. Immunotherapy of melanoma. Eur. J. Cancer Suppl. 2013, 11, 97–105.
- Choubey, D. Type I interferon (IFN)-inducible Absent in Melanoma 2 proteins in neuroinflammation: Implications for Alzheimer’s disease. J. Neuroinflam. 2019, 16, 236.
- Coventry, B.J. Therapeutic vaccination immunomodulation: Forming the basis of all cancer immunotherapy. Ther. Adv. Vaccines Immunother. 2019, 7, 2515135519862234.
- Sukari, A.; Abdallah, N.; Nagasaka, M. Unleash the power of the mighty T cells-basis of adoptive cellular therapy. Crit. Rev. Oncol. Hematol. 2019, 136, 1–12.
- Chen, C.; Gao, F.-H. Th17 Cells Paradoxical Roles in Melanoma and Potential Application in Immunotherapy. Front. Immunol. 2019, 10, 187.
- Babacan, N.A.; Eroglu, Z. Treatment Options for Advanced Melanoma After Anti-PD-1 Therapy. Curr. Oncol. Rep. 2020, 22, 38.
- Haanen, J.; Ernstoff, M.S.; Wang, Y.; Menzies, A.M.; Puzanov, I.; Grivas, P.; Larkin, J.; Peters, S.; Thompson, J.A.; Obeid, M. Autoimmune diseases and immune-checkpoint inhibitors for cancer therapy: Review of the literature and personalized risk-based prevention strategy. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2020, 31, 724–744.
- Sadozai, H.; Gruber, T.; Hunger, R.E.; Schenk, M. Recent successes and future directions in immunotherapy of cutaneous melanoma. Front. Immunol. 2017, 8, 1–25.
- Grzywa, T.M.; Paskal, W.; Włodarski, P.K. Intratumor and Intertumor Heterogeneity in Melanoma. Transl. Oncol. 2017, 10, 956–975.
- Rambow, F.; Marine, J.C.; Goding, C.R. Melanoma plasticity and phenotypic diversity: Therapeutic barriers and opportunities. Genes Dev. 2019, 33, 1295–1318.
- Vandamme, N.; Berx, G. Melanoma cells revive an embryonic transcriptional network to dictate phenotypic heterogeneity. Front. Oncol. 2014, 4, 1–6.
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28.
- Schatton, T.; Murphy, G.F.; Frank, N.Y.; Yamaura, K.; Gasser, A.M.; Gasser, M.; Zhan, Q.; Jordan, S.; Duncan, L.M.; Weishaupt, C.; et al. Identification of cells initiating human melanomas. Nature 2008, 451, 345–349.
- Büttner, R.; Longshore, J.W.; López-Ríos, F.; Merkelbach-Bruse, S.; Normanno, N.; Rouleau, E.; Penault-Llorca, F. Implementing TMB measurement in clinical practice: Considerations on assay requirements. ESMO Open 2019, 4, 1–12.
- Diener, J.; Sommer, L. Reemergence of neural crest stem cell-like states in melanoma during disease progression and treatment. Stem Cells Transl. Med. 2021, 10, 522–533.
- Quintes, S.; Brinkmann, B.G.; Ebert, M.; Fröb, F.; Kungl, T.; Arlt, F.A.; Tarabykin, V.; Huylebroeck, D.; Meijer, D.; Suter, U.; et al. Zeb2 is essential for Schwann cell differentiation, myelination and nerve repair. Nat. Neurosci. 2016, 19, 1050–1059.
- Denecker, G.; Vandamme, N.; Akay, Ö.; Koludrovic, D.; Taminau, J.; Lemeire, K.; Gheldof, A.; De Craene, B.; Van Gele, M.; Brochez, L.; et al. Identification of a ZEB2-MITF-ZEB1 transcriptional network that controls melanogenesis and melanoma progression. Cell Death Differ. 2014, 21, 1250–1261.
- Koen, E.J.; Collier, A.B. Particle-in-cell simulations of a beam driven plasma. Phys. Plasmas 2012, 4, 1420–1428.
- Lamouille, S.; Xu, J.; Derynck, R. Fakultas Psikologi Dan Sosial Budaya Universitas Islam Indonesia Yogyakarta. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196.
- Caramel, J.; Papadogeorgakis, E.; Hill, L.; Browne, G.J.; Richard, G.; Wierinckx, A.; Saldanha, G.; Sborne, J.; Hutchinson, P.; Tse, G.; et al. A Switch in the Expression of Embryonic EMT-Inducers Drives the Development of Malignant Melanoma. Cancer Cell 2013, 24, 466–480.
- Hao, L.; Ha, J.R.; Kuzel, P.; Garcia, E.; Persad, S. Cadherin switch from E- to N-cadherin in melanoma progression is regulated by the PI3K/PTEN pathway through Twist and Snail. Br. J. Dermatol. 2012, 166, 1184–1197.
- Kim, J.; Lo, L.; Dormand, E.; Anderson, D.J. SOX10 maintains multipotency and inhibits neuronal. Neuron 2003, 38, 17–31.
- Paratore, C.; Goerich, D.E.; Suter, U.; Wegner, M.; Sommer, L. Survival and glial fate acquisition of neural crest cells are regulated by an interplay between the transcription factor Sox10 and extrinsic combinatorial signaling. Development 2001, 128, 3949–3961.
- Civenni, G.; Walter, A.; Kobert, N.; Mihic-Probst, D.; Zipser, M.; Belloni, B.; Seifert, B.; Moch, H.; Dummer, R.; van den Broek, M.; et al. Human CD271-positive melanoma stem cells associated with metastasis establish tumor heterogeneity and long-term growth. Cancer Res. 2011, 71, 3098–3109.
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Ii, M.H.W.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196.
- Graham, D.K.; DeRyckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine-sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785.
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44.
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018, 174, 843–855.e19.
- Müller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.M.; Foppen, M.H.G.; et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014, 5, 5712.
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594.
- Fang, D.; Nguyen, T.K.; Leishear, K.; Finko, R.; Kulp, A.N.; Hotz, S.; Van Belle, P.A.; Xu, X.; Elder, D.E.; Herlyn, M. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005, 65, 9328–9337.
- Madjd, Z.; Erfani, E.; Gheytanchi, E.; Moradi-Lakeh, M.; Shariftabrizi, A.; Asadi-Lari, M. Expression of CD133 cancer stem cell marker in melanoma: A systematic review and meta-analysis. Int. J. Biol. Markers 2016, 31, e118–e125.
- Zabierowski, S.E.; Herlyn, M. Melanoma Stem Cells: The Dark Seed of Melanoma. J. Clin. Oncol. 2008, 26, 2890–2894.
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435.
- Fallahi-Sichani, M.; Becker, V.; Izar, B.; Baker, G.J.; Lin, J.; Boswell, S.A.; Shah, P.; Rotem, A.; Garraway, L.A.; Sorger, P.K. Adaptive resistance of melanoma cells to RAF inhibition via reversible induction of a slowly dividing de-differentiated state. Mol. Syst. Biol. 2017, 13, 905.
- Beaumont, K.; Mohana-Kumaran, N.; Haass, N. Modeling Melanoma In Vitro and In Vivo. Healthcare 2013, 2, 27–46.
- Park, E.S.; Rabinovsky, R.; Carey, M.; Hennessy, B.T.; Agarwal, R.; Liu, W.; Ju, Z.; Deng, W.; Lu, Y.; Woo, H.G.; et al. Integrative analysis of proteomic signatures, mutations, and drug responsiveness in the NCI 60 cancer cell line set. Mol. Cancer Ther. 2010, 9, 257–267.
- Caputo, E.; Maiorana, L.; Vasta, V.; Pezzino, F.M.; Sunkara, S.; Wynne, K.; Elia, G.; Marincola, F.M.; McCubrey, J.A.; Libra, M.; et al. Characterization of human melanoma cell lines and melanocytes by proteome analysis. Cell Cycle 2011, 10, 2924–2936.
- Andrique, L.; Recher, G.; Alessandri, K.; Pujol, N.; Feyeux, M.; Bon, P.; Cognet, L.; Nassoy, P.; Bikfalvi, A. A model of guided cell self-organization for rapid and spontaneous formation of functional vessels. Sci. Adv. 2019, 5, 1–12.
- Ferreira, L.P.; Gaspar, V.M.; Mano, J.F. Design of spherically structured 3D in vitro tumor models -Advances and prospects. Acta Biomater. 2018, 75, 11–34.
- Ramgolam, K.; Lauriol, J.; Lalou, C.; Lauden, L.; Michel, L.; de la Grange, P.; Khatib, A.-M.; Aoudjit, F.; Charron, D.; Alcaide-Loridan, C.; et al. Melanoma spheroids grown under neural crest cell conditions are highly plastic migratory/invasive tumor cells endowed with immunomodulator function. PLoS ONE 2011, 6, e18784.
- Schäfer, M.E.A.; Klicks, J.; Hafner, M.; Rudolf, R. Preparation, Drug Treatment, and Immunohistological Analysis of Tri-Culture Spheroid 3D Melanoma-Like Models. Methods Mol. Biol. 2021, 2265, 173–183.
- Saleh, N.A.; Rode, M.P.; Sierra, J.A.; Silva, A.H.; Miyake, J.A.; Filippin-Monteiro, F.B.; Creczynski-Pasa, T.B. Three-dimensional multicellular cell culture for anti-melanoma drug screening: Focus on tumor microenvironment. Cytotechnology 2021, 73, 35–48.
- Li, Y.; Kumacheva, E. Hydrogel microenvironments for cancer spheroid growth and drug screening. Sci. Adv. 2018, 4, eaas8998.
- Khawar, I.A.; Park, J.K.; Jung, E.S.; Lee, M.A.; Chang, S.; Kuh, H.J. Three Dimensional Mixed-Cell Spheroids Mimic Stroma-Mediated Chemoresistance and Invasive Migration in hepatocellular carcinoma. Neoplasia 2018, 20, 800–812.
- Riffle, S.; Pandey, R.N.; Albert, M.; Hegde, R.S. Linking hypoxia, DNA damage and proliferation in multicellular tumor spheroids. BMC Cancer 2017, 17, 1–12.
- Herter, S.; Morra, L.; Schlenker, R.; Sulcova, J.; Fahrni, L.; Waldhauer, I.; Lehmann, S.; Reisländer, T.; Agarkova, I.; Kelm, J.M.; et al. A novel three-dimensional heterotypic spheroid model for the assessment of the activity of cancer immunotherapy agents. Cancer Immunol. Immunother. 2017, 66, 129–140.
- Li, L.; Fukunaga-Kalabis, M.; Herlyn, M. The three-dimensional human skin reconstruct model: A tool to study normal skin and melanoma progression. J. Vis. Exp. 2011, 12, 1–5.
- Gola, M.; Czajkowski, R.; Bajek, A.; Dura, A.; Drewa, T. Melanocyte stem cells: Biology and current aspects. Med. Sci. Monit. 2012, 18, 155–159.
- Hosaka, C.; Kunisada, M.; Koyanagi-Aoi, M.; Masaki, T.; Takemori, C.; Taniguchi-Ikeda, M.; Aoi, T.; Nishigori, C. Induced pluripotent stem cell-derived melanocyte precursor cells undergoing differentiation into melanocytes. Pigment Cell Melanoma Res. 2019, 32, 623–633.
- Caputo, E.; Miceli, R.; Motti, M.L.; Taté, R.; Fratangelo, F.; Botti, G.; Mozzillo, N.; Carriero, M.V.; Cavalcanti, E.; Palmieri, G.; et al. AurkA inhibitors enhance the effects of B-RAF and MEK inhibitors in melanoma treatment. J. Transl. Med. 2014, 12, 1–9.
- Müller, I.; Kulms, D. A 3D Organotypic Melanoma Spheroid Skin Model. J. Vis. Exp. 2018, 135, e57500.
- Bartfeld, S.; Clevers, H. Organoids as model for infectious diseases: Culture of human and murine stomach organoids and microinjection of helicobacter pylori. J. Vis. Exp. 2015, 2015, 1–9.
- Leslie, J.L.; Young, V.B. A whole new ball game: Stem cell-derived epithelia in the study of host-microbe interactions. Anaerobe 2016, 37, 25–28.
- Garcez, P.P.; Loiola, E.C.; Madeiro da Costa, R.; Higa, L.M.; Trindade, P.; Delvecchio, R.; Nascimento, J.M.; Brindeiro, R.; Tanuri, A.; Rehen, S.K. Zika virus impairs growth in human neurospheres and brain organoids. Science 2016, 352, 816–818.
- Gjorevski, N.; Sachs, N.; Manfrin, A.; Giger, S.; Bragina, M.E.; Ordóñez-Morán, P.; Clevers, H.; Lutolf, M.P. Designer matrices for intestinal stem cell and organoid culture. Nature 2016, 539, 560–564.
- van Duinen, V.; Trietsch, S.J.; Joore, J.; Vulto, P.; Hankemeier, T. Microfluidic 3D cell culture: From tools to tissue models. Curr. Opin. Biotechnol. 2015, 35, 118–126.
- Doherty, E.L.; Aw, W.Y.; Hickey, A.J.; Polacheck, W.J. Microfluidic and Organ-on-a-Chip Approaches to Investigate Cellular and Microenvironmental Contributions to Cardiovascular Function and Pathology. Front. Bioeng. Biotechnol. 2021, 9, 1–14.
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772.
- Valencia, L.; Jorcano, J.L.; Velasco, D. Skin-on-a-chip models: General overview and future perspectives. APL Bioeng. 2021, 5, 030901.
- Guenat, O.T.; Berthiaume, F. Incorporating mechanical strain in organs-on-a-chip: Lung and skin. Biomicrofluidics 2018, 12, 042207.
- Jeffrey, R.; Wozniak; Edward, P.; Riley; Michael, E.; Charness, M.D. Kidney-on-a-chip: Untapped opportunities HHS Public Access. Physiol. Behav. 2019, 176, 139–148.
- Beckwitt, C.H.; Clark, A.M.; Wheeler, S.; Taylor, D.L.; Stolz, D.B.; Griffith, L.; Wells, A. Liver ‘organ on a chip. Exp. Cell Res. 2018, 363, 15–25.
- Meijer, T.G.; Jager, A.; Gent, D.C. Van Ex vivo tumor culture systems for functional drug testing and therapy response prediction-Meijer-2017. Futur. Sci. OA 2017, 3, FSO190.
- Vaira, V.; Fedele, G.; Pyne, S.; Fasoli, E.; Zadra, G.; Bailey, D.; Snyder, E.; Faversani, A.; Coggi, G.; Flavin, R.; et al. Preclinical model of organotypic culture for pharmacodynamic profiling of human tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 8352–8356.
- Bougherara, H.; Mansuet-Lupo, A.; Alifano, M.; Ngô, C.; Damotte, D.; Le Frère-Belda, M.A.; Donnadieu, E.; Peranzoni, E. Real-time imaging of resident T cells in human lung and ovarian carcinomas reveals how different tumor microenvironments control T lymphocyte migration. Front. Immunol. 2015, 6, 1–12.
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guérin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti–PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050.
More