Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ahmed M. Gouda | + 893 word(s) | 893 | 2021-11-08 07:08:32 | | | |
| 2 | Conner Chen | Meta information modification | 893 | 2021-11-25 02:33:15 | | | | |
| 3 | Conner Chen | Meta information modification | 893 | 2021-11-25 02:33:38 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Gouda, A. Classification of the Approved EGFR-TKIs. Encyclopedia. Available online: https://encyclopedia.pub/entry/16363 (accessed on 07 August 2026).
Gouda A. Classification of the Approved EGFR-TKIs. Encyclopedia. Available at: https://encyclopedia.pub/entry/16363. Accessed August 07, 2026.
Gouda, Ahmed. "Classification of the Approved EGFR-TKIs" Encyclopedia, https://encyclopedia.pub/entry/16363 (accessed August 07, 2026).
Gouda, A. (2021, November 24). Classification of the Approved EGFR-TKIs. In Encyclopedia. https://encyclopedia.pub/entry/16363
Gouda, Ahmed. "Classification of the Approved EGFR-TKIs." Encyclopedia. Web. 24 November, 2021.
Copy Citation
Targeting EGFR with small-molecule inhibitors is a valid strategy in cancer therapy. Since the approval of the first EGFR-TKI in 2003, a huge number of EGFR inhibitors were reported. Classification of these inhibitors could help the researchers to understand their structure-activity relationship. Herein, we introduce different types of classifications of the EGFR-TKIs, which received global approval for clinical use. In the following, the EGFR-targeting drugs are classified based on their chemistry, clinical use, target kinases, and the type of inhibition/interaction with EGFR.
kinase inhibitor
EGFR
classification of EGFR-TKIs
chemical classification
Pharmacological classification
target kinase-based classification
Type of inhibition/interaction-based classification
1. Chemical Classification
Based on their chemical structures, the approved EGFR-TKIs can be classified into three subclasses. The first subclass is the aminopyrimidine derivatives which include almonertinib, brigatinib, and osimertinib. In this study, olmutinib is considered as a fused aminopyrimidine derivative. The second group has a quinoline-4,6-diamine nucleus and includes two derivatives, neratinib and pyrotinib. The third group is the quinazoline-4,6-diamine-based derivatives which include afatinib, dacomitinib, erlotinib, icotinib, lapatinib, simotinib, and vandetanib, as shown in Figure 4.
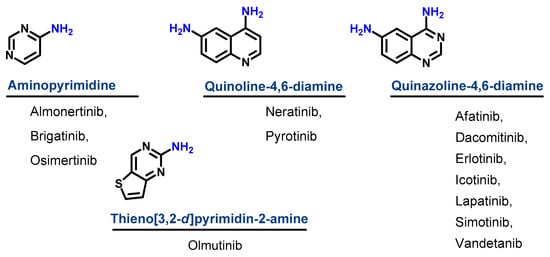
Figure 1. Chemical classification of EGFR-TKIs.
2. Classification Based on the Types of Interaction with EGFR
Based on the type of inhibition of the EGFR activity, the approved EGFR-TKIs can also be classified as reversible and irreversible inhibitors, as shown in Figure 5. For the first type, the inhibitors competitively bind to the ATP binding site in the EGFR through noncovalent interactions involving electrostatic, hydrogen-bonding, and hydrophobic interactions [1].
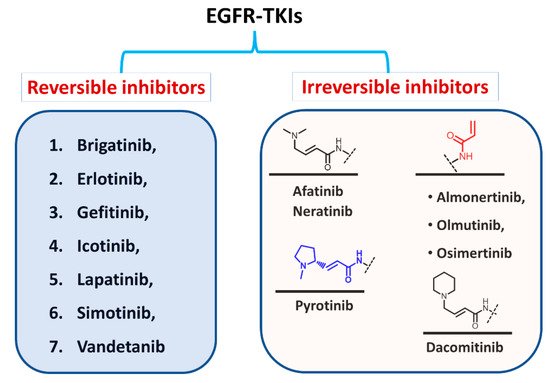
Figure 2. Classification of EGFR-TKIs based on the nature of inhibition of EGFR.
In the second type, the EGFR inhibitors form a covalent bond with the cysteine residue in the EGFR. The structure of these inhibitors is characterized by the presence of an electrophilic side chain that acts as a Michael acceptor which chemically reacts with the cysteine thiol group to form a covalent adduct [2].
3. Classification Based on the Clinical Use
The EGFR inhibitors can also be classified into three groups based on the types of cancers for which they were approved [3]. The first group includes drugs that were approved for the treatment of NSCLC such as afatinib, almonertinib, brigatinib, dacomitinib, erlotinib, gefitinib, icotinib, olmutinib, and osimertinib. The second type includes drugs approved for the treatment of breast cancer such as lapatinib and neratinib, and pyrotinib. On the other hand, the third group includes drugs approved for other types of cancers such as thyroid cancer (vandetanib), pancreatic cancer (erlotinib), and solid cancers (simotinib).
4. Classification Based on the Target Kinases
The approved EGFR-TKIs may also be classified according to their target kinases into (1) selective EGFR inhibitors which target EGFR with high selectivity such as gefitinib; (2) dual EGFR inhibitors such as lapatinib which can target EGFR and ErbB-2; and (3) multi-kinase inhibitors such as brigatinib, pyrotinib, and vandetanib. These drugs have broad-spectrum activity against multiple kinases other than the EGFR [4].
5. Classification into the First, Second, and Third Generation
Among the approved EGFR-TKIs, gefitinib, erlotinib, lapatinib, and icotinib are classified as first-generation EGFR inhibitors, as shown in Figure 6. The drugs in this class bind reversibly to the PTK domain of the EGFR, which leads to the inhibition of the binding of ATP to the EGFR and consequently to the inhibition of EGFR activation and cellular proliferation [5]. The chemical structures of the four drugs consist of a basic quinazoline nucleus attached to a substituted aniline moiety, as shown in Figure 3.
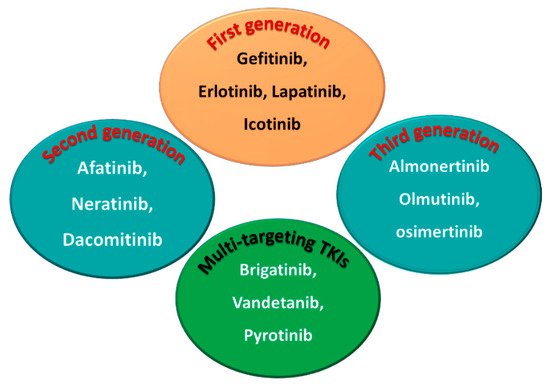
Figure 3. First-, second-, and third-generation EGFR-TKIs and the multi-targeting inhibitors.
On the other hand, afatinib, neratinib, and dacomitinib are classified as second-generation EGFR-TKIs. These three drugs contain a Michael acceptor site which allows them to bind covalently to the EGFR, leading to the irreversible inhibition of the kinase activity, which provides an advantage over first-generation EGFR-TKIs [6]. The chemical structures of these drugs consist of quinazoline or a quinoline nucleus which bears a crotonamide side chain (Michael acceptor site) substituted at the terminal carbon by a tert-amino group.
The third-generation EGFR inhibitors include almonertinib, olmutinib, and osimertinib [7]. The chemical structures of these drugs include a pyrimidine nucleus attached to a substituted anilino or phenoxy moiety. These moieties bear an acrylamide group that, as noted above, can form a covalent bond with the cysteine residue in the EGFR.
On the other hand, brigatinib, vandetanib, and pyrotinib are classified as multi-kinase inhibitors due to their inhibitory activities against kinases other than the EGFR [8].
The emergence of EGFR T790M and C797S mutation has led to the rapid development of resistance to the first- second- and third-generation EGFR-TKIs [7]. The C797S mutation also deprives the irreversible inhibitors of the EGFR of the ability to bind covalently to the kinase. In addition, the emergence of EGFR double and triple (del19/L858R + C797S ± T790M) mutations challenges the therapeutic effectiveness of EGFR-TKIs. These problems underscore the ongoing need to develop new and potent EGFR-TKIs.
Recently, several fourth-generation allosteric EGFR inhibitors that bind to a site in the EGFR other than the PTK domain were reported [9]. However, although these inhibitors are ineffective against NSCLC with a mutated EGFR, they display synergistic anticancer effects when combined with an EGFR inhibitor such as osimertinib or the monoclonal antibody, cetuximab.
Currently, several fourth-generation EGFR-TKIs are being evaluated in regard to their efficacy against cancers carrying double or triple EGFR mutations [10][11][12]. One of these compounds, BBT-176, is in Phase I/II trials in NSCLC patients with advanced lung cancer [12], but these inhibitors are not yet approved for clinical use.
References
- Akher, F.B.; Farrokhzadeh, A.; Soliman, M.E.S. Covalent vs. Non-Covalent Inhibition: Tackling Drug Resistance in EGFR—A Thorough Dynamic Perspective. Chem. Biodivers. 2019, 16, e1800518.
- Hossam, M.; Lasheen, D.S.; Abouzid, K.A.M. Covalent EGFR Inhibitors: Binding Mechanisms, Synthetic Approaches, and Clinical Profiles. Arch. Pharm. (Weinh.) 2016, 349, 573–593.
- Uribe, M.L.; Marrocco, I.; Yarden, Y. EGFR in Cancer: Signaling Mechanisms, Drugs, and Acquired Resistance. Cancers 2021, 13, 2748.
- You, K.S.; Yi, Y.W.; Cho, J.; Park, J.-S.; Seong, Y.-S. Potentiating Therapeutic Effects of Epidermal Growth Factor Receptor Inhibition in Triple-Negative Breast Cancer. Pharmaceuticals 2021, 14, 589.
- Dutta, P.R.; Maity, A. Cellular responses to EGFR inhibitors and their relevance to cancer therapy. Cancer Lett. 2007, 254, 165–177.
- Stasi, I.; Cappuzzo, F. Second generation tyrosine kinase inhibitors for the treatment of metastatic non-small-cell lung cancer. Transl. Respir. Med. 2014, 2, 2.
- Nagasaka, M.; Zhu, V.W.; Lim, S.M.; Greco, M.; Wu, F.; Ou, S.-H.I. Beyond Osimertinib: The Development of Third-Generation EGFR Tyrosine Kinase Inhibitors for Advanced EGFR+ NSCLC. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2021, 16, 740–763.
- Zhang, C.; Leighl, N.B.; Wu, Y.-L.; Zhong, W.-Z. Emerging therapies for non-small cell lung cancer. J. Hematol. Oncol. 2019, 12, 45.
- Tripathi, S.K.; Biswal, B.K. Allosteric mutant-selective fourth-generation EGFR inhibitors as an efficient combination therapeutic in the treatment of non-small cell lung carcinoma. Drug Discov. Today 2021, 26, 1466–1472.
- Wang, S.; Song, Y.; Liu, D. EAI045: The fourth-generation EGFR inhibitor overcoming T790M and C797S resistance. Cancer Lett. 2017, 385, 51–54.
- Du, X.; Yang, B.; An, Q.; Assaraf, Y.G.; Cao, X.; Xia, J. Acquired resistance to third-generation EGFR-TKIs and emerging next-generation EGFR inhibitors. Innovations 2021, 2, 100103.
- Piper-Vallillo, A.J.; Sequist, L.V.; Piotrowska, Z. Emerging Treatment Paradigms for EGFR-Mutant Lung Cancers Progressing on Osimertinib: A Review. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 2926–2936.
More
Information
Subjects:
Chemistry, Medicinal
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
6.0K
Entry Collection:
Organic Synthesis
Revisions:
3 times
(View History)
Update Date:
25 Nov 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No