Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Enrico M. Clini | + 3532 word(s) | 3532 | 2021-11-17 01:49:14 | | | |
| 2 | Bruce Ren | Meta information modification | 3532 | 2021-11-24 09:06:32 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Clini, E.M. Molecular Mechanisms from Lung Fibrosis to Lung Cancer. Encyclopedia. Available online: https://encyclopedia.pub/entry/16333 (accessed on 24 July 2026).
Clini EM. Molecular Mechanisms from Lung Fibrosis to Lung Cancer. Encyclopedia. Available at: https://encyclopedia.pub/entry/16333. Accessed July 24, 2026.
Clini, Enrico M.. "Molecular Mechanisms from Lung Fibrosis to Lung Cancer" Encyclopedia, https://encyclopedia.pub/entry/16333 (accessed July 24, 2026).
Clini, E.M. (2021, November 24). Molecular Mechanisms from Lung Fibrosis to Lung Cancer. In Encyclopedia. https://encyclopedia.pub/entry/16333
Clini, Enrico M.. "Molecular Mechanisms from Lung Fibrosis to Lung Cancer." Encyclopedia. Web. 24 November, 2021.
Copy Citation
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, fibrosing interstitial lung disease (ILD) of unknown aetiology, with a median survival of 2–4 years from the time of diagnosis. Although IPF has unknown aetiology by definition, there have been identified several risks factors increasing the probability of the onset and progression of the disease in IPF patients such as cigarette smoking and environmental risk factors. Cigarette smoking together with concomitant emphysema might predispose IPF patients to lung cancer (LC), mostly to non-small cell lung cancer (NSCLC), increasing the risk of cancer development.
idiopathic pulmonary fibrosis
lung cancer
myofibroblast
cancer associated fibroblasts (CAFs)
mechanotrasduction
1. Introduction
Interstitial lung diseases represent a broad spectrum of lung pathologies that affect the lung parenchyma causing diffuse inflammation and fibrosis [1]. Among them, idiopathic pulmonary fibrosis (IPF) is a rare lung disease with an unknown cause and a median survival of 2–4 years from the time of diagnosis. The onset and progression of IPF leads to massive changes in the architecture of lungs and their biomechanical properties that often culminate in respiratory failure due to the impairment of alveolar gas-exchange and the decline of lung functions [2][3]. Although the diagnosis of IPF can be extremely challenging due to the heterogenous nature of this disease [4], it is recognized by clinicopathological criteria, including the radiographic and histological hallmarks pattern of usual interstitial pneumonia (UIP) [5]. Currently, there are two antifibrotic drugs used as a therapeutic strategy for IPF patients, Pirfenidone and Nintedanib, that are able to slow down the respiratory functional decline and improve survival in IPF patients [6][7][8]. Despite this, IPF still has a high mortality rate and the survival times are quite heterogenous [9][10][11]. Although IPF is idiopathic by definition and is not classified as a genetic disease, there are both environmental and genetic risk factors (or “genetic susceptibility”) that can play a fundamental role in the initiation and progression of IPF [12][13]. Among them, the most studied and validated genetic risk factor is represented by the single nucleotide polymorphism in the promoter region of the mucin 5B (MUC5B) responsible for sporadic and familial IPF [12]. Furthermore, among the environmental risks there are exposure to cigarette smoke and inhalation of wood and metal dust, which might severely affect “genetically susceptible” patients [14] with the resulting alteration in the regulation of key genes contributing to the pathogenesis of IPF. Several studies have demonstrated that viral infection, including with the recently prominent SARS-CoV-2, might be responsible for the initiation or exacerbation of pulmonary fibrosis. In particular, it has been postulated that the “cytokine storm” caused by the exaggerated inflammatory response following SARS-CoV-2 infection, as well as micro-thrombotic hypotheses, may predispose patients with COVID-19 pneumonia to aberrant mechanisms of repair and fibrosis [15] culminating in acute lung injury and interstitial lung disease [16]. Although different prospective studies aiming to investigate the long-term pulmonary consequences of COVID-19 are still ongoing, it has been observed in the study of Wu et al. that about 40% of 201 patients with COVID-19 pneumonia developed acute respiratory distress syndrome (ARDS) [17]. Indeed, like SARS-CoV-2, the other two known coronaviruses, both the severe acute respiratory syndrome coronavirus (SARS-CoV; SARS) and Middle East respiratory syndrome coronavirus (MERS-CoV; MERS), caused in some patients interstitial abnormalities and lung functional decline. [18][19].
In the past few years, there has been growing interest in the role of comorbidities in IPF study. Smoking history, elderly age, male sex, and emphysema might represent strong risk factors for developing lung cancer (LC) in IPF patients; thus, besides the concomitant risk factors, IPF itself might be considered a risk factor for lung carcinogenesis [20][21][22]. Lung cancer is the most diffuse cause of cancer death worldwide. It has been estimated that about 85% of LC patients have a diagnosis of non-small cell lung cancer (NSLC). In the majority of patients, the onset of LC is related to tobacco smoking [23] (Figure 1).
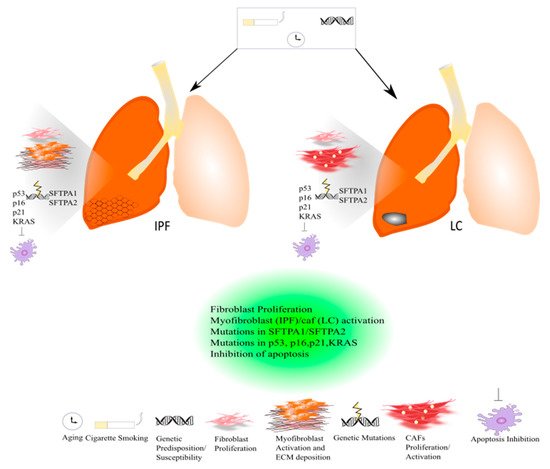
Figure 1. Common pathogenic mechanisms between lung cancer and IPF.
Recent studies have focused on the identification of common molecular pathways between IPF and LC for a better therapeutic strategy and optimal management of patients with both diseases. There are many common cellular and molecular mechanisms that might predispose patients to the onset and development of IPF and LC such as the activation and proliferation of both myofibroblasts (IPF) and cancer-associated fibroblasts (CAFs), and the alteration of growth factors expression level [24]. Myofibroblasts represent the cellular key players in IPF since their proliferation and activation under profibrotic stimuli lead to the secretion and deposition of extracellular matrix (ECM) proteins, which are responsible for causing fibrosis [25]. Thus, this process culminates in increased lung structural rigidity that compromises the lung’s biomechanical properties and thickens of the alveolar–capillary barrier with aberrant alveolar gas exchange function. According to recent investigations, the myofibroblasts derive from different lung resident cell populations such as the interstitial lung fibroblasts, the lipofibroblasts [26], lung resident mesenchymal stromal cells (LR-MSCs) [27][28], the perycites [29] and mesothelial cells [30], ruling out the contribution of both epithelial cells in the so-called epithelial mesenchymal transition (EMT) [31] and circulating fibrocytes [32]. In LC, cancer-associated fibroblasts (CAFs) are the cellular key players, like myofibroblasts in IPF pathogenesis [33]. Several studies have been focused on the identification of the origins, biological characteristics, and the role of CAFs as the major component of the stroma during carcinogenesis. Intriguingly, among the components of the tumour microenvironment (TME), CAFs play a fundamental role in drug resistance in NSCLC, protecting tumours from the effects of chemotherapeutic drugs [34][35]. As is the case with IPF, there are several hypotheses still under debate regarding the cellular sources of CAFs in tumours; among them are (1) the resident fibroblasts, which can differentiate to CAFs during the course of tumour progression under the orchestration of specific signalling pathways; (2) cancer associated adipocytes (CAAs) that are present in tumour stroma and might derive from circulating progenitors in the bone marrow [36]; (3) bone-marrow-derived mesenchymal stromal cells (BM-MSCs) and hematopoietic stem cells (HSCs) [37]; (4) epithelial cells through the epithelial mesenchymal transition (EMT) [38]; (5) vascular endothelial cells through the endothelial–mesenchymal transition (EndMT), crucial for tumour angiogenesis [39] (Figure 2) Thus, CAFs exhibit high heterogeneity in terms of origin as well as surface markers and resident organs. During malignancy while the tumour progresses, CAFs contribute to promote tumour growth, metastasis, and drug resistance. Here, tumor cells together with non-malignant stromal cells trigger CAF activation through inflammatory mediators such as transforming growth factor beta (TGF-β), interleukin (IL)-1, and interleukin (IL)-6 that contribute to inflammation and carcinogenesis [40][41]. To this purpose, TGF-β that can be considered as a master molecular regulator of profibrotic signaling, promoting lung cancer progression and triggering mitogenic stimuli to lung cancer cells. Furthermore, among the common signalling pathways characterizing both IPF and LC progression, there is the Wnt/β-catenin pathway that has been involved both in cancer progression and the EMT process through its target genes, cyclin-D1 and matrix metalloproteinase (MMP)-7, contributing to the pathogenesis of IPF [42]. Aberrant activation of phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) causes cancer invasion and the progression of lung fibrosis [43] with the activation of profibrotic downstream signalling mediators such as TGF-β1 and platelet-derived growth factor (PDGF). The sonic hedgehog (Shh) pathway is also activated both in bronchial epithelial cells of honeycomb cysts and in cancer fibroblasts, being responsible for resistance to fibroblast apoptosis, tumour growth, metastasis, and chemotherapy resistance [44]. (Figure 2) Furthermore, cellular senescence is associated with the progression of pulmonary fibrosis through different mechanisms and central players in the lung niche. Among them, Wiley et al. [45] demonstrated that the biologically active profibrotic lipids, namely the leukotrienes (LT), are involved in the senescence-associated secretory phenotype (SASP) which represents one of the mechanisms responsible for the progression of pulmonary fibrosis. They demonstrated that the LT-rich conditioned medium (CM) of senescent lung fibroblasts triggered profibrotic signalling in modified fibroblasts treated with inhibitors of ALOX5, the main enzyme in LT biosynthesis [45]. To this purpose, another recent study from Li et al. [46], stated that blocking the biosynthesis of leukotriene B4 (LTB4) would be an efficient therapeutic strategy in the treatment of both IPF and acute lung injury (ALI). They performed in vivo experiments, where they observed a decreased neutrophilic inflammation in an IPF mouse model at early stage, as well as decreased LPS-induced ALI through LTB4 blocking biosynthesis in vivo. Indeed, several works have been published about the role of LT in lung cancer progression. Among them, Poczobutt et al. [47] showed a selective production of leukotrienes by inflammatory cells of the microenvironment during lung cancer progression through an orthotopic model of lung cancer progression and by liquid chromatograph coupled with tandem mass spectrometry (LC/MS/MS) [47].
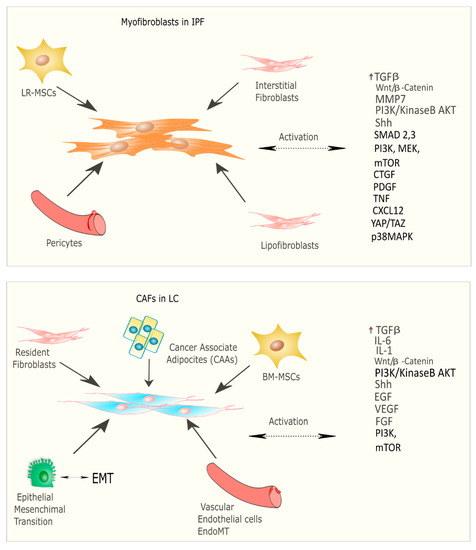
Figure 2. Origin and signalling pathway leading to the activation of myofibroblasts in IPF and CAFs in LC.
Indeed, there are also common genetic mechanisms shared by both diseases based on mutations in surfactant protein genes (SFTPA2-A1) that lead to impaired protein secretion, endoplasmic reticular stress, and apoptosis, culminating in the onset and progression of IPF or adenocarcinoma [24]. (Figure 1) To date, the approved IPF therapies, Pirfenidone and Nintedanib, are also active in LC. In particular, Nintedanib is approved as a treatment in NSCLC, and Pirfenidone has shown anti-neoplastic effects in preclinical studies [24].
2. Diagnostic Approaches for Idiopathic Pulmonary Fibrosis and Lung Cancer
Idiopathic pulmonary fibrosis (IPF) is a rare pulmonary disease with an incidence ranging from 0.09 to 0.49 in Europe [48]. Among these patients, there is an increased risk of developing lung cancer (LC), with a relative prevalence reported from 2.7% to 48% [49][50]. The correlation between IPF and lung cancer is still being debated [6], although recent studies have demonstrated possible connections [49][50]. In particular, Ozawa et al. [51] showed an increased incidence of lung cancer in a retrospective study of 103 patients with IPF. Similar results have been described by Tomassetti et al. [52], with cancer occurring in 30% of studied patients. One of the most interesting approaches was to analyze the period of onset of lung cancer in IPF-LC patients. In 2021, Alomaish H. et al. [53] reported a LC incidence of 13,5% IPF patients. Because lung cancer has a high incidence in patients with IPF having an important impact on the survival of these patients, the scientific community has focused its attention on identifying predictive factors for lung cancer in IPF patients [54][55][56]. These characteristics are mainly described in older patients with IPF at diagnosis who have a history of smoking and emphysema [51][56][57]. However, the causes inducing lung cancer in this population of patients have not been clarified yet [58][59]. One of the most interesting aspects, which seems important to consider, is a rapid annual decline of forced vital capacity (FVC) as a lung-cancer-predisposing factor [60]. In particular, it seems that tissue damage and abnormal repair is the key to the connections between IPF and LC [59]. Indeed, it has been thought that patients with a very rapid decrease in FVC and consequent IPF progression are more sensitive to lung cancer development; however, further studies will need to be conducted to clarify the common factors between cancer and IPF. A recent study demonstrated a median time from IPF to lung cancer of 38 months [53], although Tomassetti et al. [52] found the time to be around 30 months. Besides the decrease in FVC and carbon monoxide diffusion capacity (DLCO) in patients who developed both IPF and LC, another aspect to investigate are the histopathological findings. Previous studies have shown that squamous cell carcinoma is the most common histologic type encountered in IPF [51][52][53][54][55][56][57][58][59][60][61][62]. This aspect would highlight the importance of considering older patients with a diagnosis of IPF who have a long and heavy history of smoking and whose median FVC and DLCO are slightly lower than normal [63]. In particular, radiological findings are crucial to consider since round or ovoid masses are frequently observed [64][65] in correspondance to the lower lobes [64][66]. The recent guidelines recommend an annual low-dose chest CT scan in high-risk patients [67][68], but there are no recommendations for IPF patients. It is probably important to consider screening patients with IPF, especially in the first two years after diagnosis, with an annual or shorter-term chest CT scan to follow the possible presence and evolution of lung cancer nodules [69]. Since recent studies have described a worse survival rate in patients with IPF and lung cancer [70][71], it would be fundamental to establish a surveillance protocol in order to set a diagnosis and medical treatment for these patients.
The scientific community believes that multidisciplinary approaches are the diagnostic gold standard for patients with moderate IPF and IPF-LC [72][73]. In particular, in patients with IPF of mild to moderate functional impairment (FVC > 50%, DLCO > 35%) and the association of an early-stage lung cancer or metastasis, the common approaches are surgery for the early stages and stereotactic radiotherapy for the advanced stages, together with anti-fibrotic treatments used for more than 50% of patients [74][75][76]. In critical patients with advanced IPF and operable lung cancer but impaired pulmonary function, the recommended treatments are anti-fibrotics, immune checkpoint inhibitors, and targeted therapy [73][77]. Another interesting aspect for diagnostic and therapeutic approaches is the identification of biomarkers, which represents the first step toward future personalized therapies for IPF [73][77]. These approaches may have an important impact in either preventing or monitoring lung cancer in IPF patients. To this purpose, in the last decades, several potential biomarkers have been discovered [78][79].
2.1. Diagnostic Biomarkers
The most studied biomarkers able to discriminate IPF from healthy donors are the markers Krebs von den Lungen (KL)-6 [80][81][82][83], the chitinase-like protein (YKL40) [84][85], leucocytes and circulating innate immune cells [78][79][80], and surfactant proteins (SP)-A, -B, -D [86][87][88][89][90][91]. Indeed, the matrix metalloproteinase (MMP7), at higher values, seems to show a higher risk of possible interstitial lung diseases [92]. In particular, MMP7 and MMP1 are important for making a differential diagnosis between IPF and hypersensitivity pneumonitis. The recent PROFILE (Prospective Study of Fibrosis in Lung Endpoints) study that analyzed the molecular profile of more than 100 serum proteins showed a significantly higher level of MMP1, MMP7, and SP-D in IPF patients compared with healthy people. Furthermore, it showed that oncostatin M and cytokeratin 19 fragments (CYFRA-21-1) were markers for IPF patients [54][93][94][95][96][97][98]. These biomarkers may be an important aspect to investigate in the future even for lung cancer. However, the settings of serum biomarkers for clinical perspectives for IPF monitoring and further lung cancer development are still lacking and have not been considered for diagnostic purposes in IPF [99].
2.2. Prognostic Biomarkers
Functional decline and respiratory function monitoring are the best approaches at the moment for setting and monitoring IPF progression [96][97], but the possibility has recently arisen of identifying biomarkers with prognostic purposes for IPF. Although the heterogeneity of IPF patients complicates the definition of a univocal approach, a composite scoring system incorporating lung physiology, sex, and age (GAP) is more accurate at predicting mortality [98][99]. These biomarkers seem to improve prognosis for IPF patients [100][101][102][103][104][105][106][107][108][109][110][111][112][113][114]. In particular, a high concentration of MMP7 correlates with IPF severity for the decline of lung function as well as worse survival of IPF [115]. For example, in the PROFILE study, the protein MMP1/8 (CRPM) indicates pulmonary disease progression and very poor overall survival [116]. Another interesting predictor for IPF as well as lung cancer seems to be surfactant protein D and the cancer antigen (CA)19-9 [117].
2.3. Radiological Biomarkers
As previously described, several studies defined the high-resolution computed tomography (HRCT) patterns as prospective prognostic biomarkers [118][119]. In particular, a recent study claims that the employment of specific CT-associated tool such as data-driven textural analysis (DTA) [120] associated with a visual and functional changes score, might be useful in predicting IPF progression [120]. However, the definition of a unique method and score set using HRCT characteristics associated with pulmonary function test results is still lacking. Recently, Jacob et al. [121] reported that a computer score set on the quantification of parenchymal patterns including vessel-related structure was able to predict IPF mortality and functional decline, representing a potential non-invasive method for estimating gas-exchange impairment in IPF [121][122].
In summary, keeping in mind the variability and uncertainty of the approaches, the best strategies that need to be considered in asymptomatic middle-aged smokers are: (1) a chest low-dose HRCT or regular CT conducted annually; (2) optimal selection of patients for surgical lung interventions, chemotherapy, and radiotherapy; (3) the role of anti-fibrotics in preventing and treating lung cancer and reducing acute exacerbations of IPF post-operatively; (4) the settings of new molecular biomarkers that may be useful as diagnostic and predictive factors for optimal monitoring of IPF. All these aspects will be better addressed through the setting of a future consensus statement for diagnosis and management of these patients in order to standardize the diagnostic and prognostic approaches as well as also develop more focused medical treatments.
3. Common Pathogenic Mechanisms between LC and IPF: Genetic and Epigenetic Alterations in Focus
Several studies going inside the pathogenesis of IPF and LC exhibit great similarity between both diseases concerning the abnormal activation of the signalling pathway, the cellular responses, the activation of lung fibroblasts and their proliferation. To date, there is a growing interest in the epigenetic and genetic abnormalities characterizing IPF and LC that might explain the concomitant manifestation of the two diseases. Although IPF is not considered a genetic disease, genome-wide association analysis (GWA) identified different genetic variants that account for almost 30% of IPF patients that together with the environmental risks play a crucial role in both sporadic and familial IPF [13][123][124]. Among these, the most studied genetic risk factor for both familial and sporadic IPF is the single nucleotide polymorphism rs35705950 in the promoter region of the mucin 5B (MUC5B) gene [12][125]. Normally, in the alveolar epithelium, MUC5B plays a pivotal role together with mucin 5AC (MUC5AC) in the muco-ciliary clearance (MCC), removing inhaled debris and pathogens and contributing to the maintenance of the overall lung homeostasis [126]. To this purpose, it has been shown that the overexpression of full-length murine MUC5B in the AECII of two lines of C57BL/6 mice compromised the muco-ciliary clearance activity, leading to an increase in lung fibrosis of bleomycin-treated mice [1]. Moreover, other genetic variants associated with sporadic and familial IPF are represented by: genes involved in cell–cell adhesion (DSP, DPP9) fundamental in the maintenance of epithelial integrity, genes involved in innate and adaptive immune response (Toll-like receptor signalling, TOLLIP, TLR3), surfactant protein genes (SFTPA2-A1), cytokines and growth factors (IL1RN, IL8, IL4, TGF-β1), genes involved in telomere maintenance (TERT, OBFC1), and cell-cycle regulation genes (KIF15, MAD1L1, CDKN1A, TP53) [127].
Different IPF-related genetic variants have been associated with the risk to develop lung cancer. Among these, there are mutations in surfactant protein genes (SFTPA1, SFTPA2,) that have been studied in lung adenocarcinoma that compromise protein secretion and promote endoplasmic reticular stress and apoptosis [128][129]. In familial IPF, genetic variants of telomerase complex components, such as TERT (telomerase reverse transcriptase) and TERC (telomerase RNA component), lead to shortened telomeres following by genomic instability [130]. To this purpose, several studies suggest a different role of telomerase in IPF and LC since it was found that the expression level of TERT and TERC was significantly lower in the lung tissue of IPF patients compared to NSCLC tissues and controls [131]. Indeed, mutations in the p53 gene that lead to a decrease in the apoptotic process together with mutations in p16, p21, and the Kirsten rat sarcoma virus gene (KRAS) have been found both in IPF and LC [132]. (Figure 1). Furthermore, Maher et al. performed the PROFILE study of a large cohort of IPF patients, four serum biomarkers predictive of disease progression, among which are the cancer-related genes CA-19 and CA-125 [133]. In addition, Allen et al. assessed the genomic profiles of IPF-LC using targeted exome sequencing where they found several somatic mutations, among which were the TP53 and BRAF genes, which were significantly mutated in IPF-LC [134]. Intriguingly, it has been demonstrated that epidermal growth factor receptor (EGFR) and anaplastic lymphoma kinase (ALK) mutations that normally drive the therapeutic decisions in the management of LC patients are either less frequent in IPF-LC adenocarcinoma patients compared with LC adenocarcinoma patients or have not been studied, respectively [135]. The enviromental risk factors, smoking exposure and aging, that might charaterize both IPF and LC patients might induce epigenetic responses such as changes in the methylation patterns that are quite similar between both diseases according to genome-wide methylation analysis [136]. Finally, recent studies show that IPF and LC share the aberrant expression of some microRNA. Among them, miR-21 was both upregulated in patients with IPF, [137] and correlated with poor prognosis in NSCLC patients according to the meta-analysis performed [138]. Finally, miR-29, miR-200, and let-7d were found to be downregulated both in the lung tissue of IPF patients and LC tissues [139][140]. However, it is important to mention that LC is endowed with metastatic potential, disseminating around the body (especially into the brain and bone), unlike IPF, which remains still localized to the lung.
Cigarette smoking together with genetic susceptibility/predisposition and aging can lead to the onset and progression of IPF and LC. Both diseases are characterized by common and similar pathogenic mechanisms. Both in IPF and lung cancer, a massive proliferation of lung resident fibroblasts occurs that contributes to the progression of IPF and lung cancer. In IPF, activated myofibroblasts promote the deposition of ECM leading to an acute exacerbation of the disease, while in lung cancer activated cancer-associated fibroblasts (CAFs) drive the carcinogenesis. Indeed, genetic mutations in surfactant proteins such as SFTPA1 and SFTPA2 have been studied both in familial IPF and lung cancer. Genetic mutations in the p53 gene that lead to a decrease in apoptosis and p16, p21, and KRAS have been found both in IPF and LC.
References
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of idiopathic pulmonary fibrosis An Official ATS/ERS/JRS/ALAT Clinical practice guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68.
- Chanda, D.; Otoupalova, E.; Smith, S.R.; Volckaert, T.; De Langhe, S.P.; Thannickal, V.J. Developmental pathways in the pathogenesis of lung fibrosis. Mol. Asp. Med. 2019, 65, 56–69.
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952.
- Samarelli, A.V.; Tonelli, R.; Marchioni, A.; Bruzzi, G.; Gozzi, F.; Andrisani, D.; Castaniere, I.; Manicardi, L.; Moretti, A.; Tabbì, L.; et al. Fibrotic idiopathic interstitial lung disease: The molecular and cellular key players. Int. J. Mol. Sci. 2021, 22, 8952.
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824.
- Guenther, A.; Krauss, E.; Tello, S.; Wagner, J.; Paul, B.; Kuhn, S.; Maurer, O.; Heinemann, S.; Costabel, U.; Barbero, M.A.N.; et al. The European IPF registry (eurIPFreg): Baseline characteristics and survival of patients with idiopathic pulmonary fibrosis. Respir. Res. 2018, 19, 1–10.
- Crestani, B.; Huggins, J.T.; Kaye, M.; Costabel, U.; Glaspole, I.; Ogura, T.; Song, J.W.; Stansen, W.; Quaresma, M.; Stowasser, S.; et al. Long-term safety and tolerability of Nintedanib in patients with idiopathic pulmonary fibrosis: Results from the open-label extension study, INPULSIS-ON. Lancet Respir. Med. 2019, 7, 60–68.
- Cottin, V.; Koschel, D.; Günther, A.; Albera, C.; Azuma, A.; Sköld, C.M.; Tomassetti, S.; Hormel, P.; Stauffer, J.L.; Strombom, I.; et al. Long-term safety of Pirfenidone: Results of the prospective, observational PASSPORT study. ERJ Open Res. 2018, 4, 00084–02018.
- Behr, J.; Prasse, A.; Kreuter, M.; Johow, J.; Rabe, K.F.; Bonella, F.; Bonnet, R.; Grohe, C.; Held, M.; Wilkens, H.; et al. Pirfenidone in patients with progressive fibrotic interstitial lung diseases other than idiopathic pulmonary fibrosis (RELIEF): A double-blind, randomised, placebo-controlled, phase 2b trial. Lancet Respir. Med. 2021, 9, 476–486.
- Sanders, J.L.; Putman, R.K.; Dupuis, J.; Xu, H.; Murabito, J.M.; Araki, T.; Nishino, M.; Benjamin, E.J.; Levy, D.L.; Ramachandran, V.S.; et al. The Association of Aging Biomarkers, Interstitial Lung Abnormalities, and Mortality. Am. J. Respir. Crit. Care Med. 2021, 203, 1149–1157.
- Cerri, S.; Monari, M.; Guerrieri, A.; Donatelli, P.; Bassi, I.; Garuti, M.; Luppi, F.; Betti, S.; Bandelli, G.; Carpano, M.; et al. Real-life comparison of Pirfenidone and Nintedanib in patients with idiopathic pulmonary fibrosis: A 24-month assessment. Respir. Med. 2019, 159, 105803.
- Noth, I.; Zhang, Y.; Ma, S.F.; Flores, C.; Barber, M.; Huang, Y.; Broderick, S.M.; Wade, M.S.; Hysi, P.; Scuirba, J.; et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: A genome-wide association study. Lancet Respir Med. 2013, 1, 309–317.
- Hobbs, B.D.; Putman, R.K.; Araki, T.; Nishino, M.; Gudmundsson, G.; Gudnason, V.; Eiriksdottir, G.; Zilhao Nogueira, N.R.; Dupuis, J.; Xu, H.; et al. Overlap of Genetic Risk between Interstitial Lung Abnormalities and Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 200, 1402–1413.
- Abramson, M.J.; Walters, E.H. Mapping air pollution and idiopathic pulmonary fibrosis. Respirology 2021, 26, 292–293.
- Tang, X.; Du, R.H.; Wang, R.; Cao, T.Z.; Guan, L.L.; Yang, C.Q.; Zhu, Q.; Hu, M.; Li, X.Y.; Li, Y.; et al. Comparison of Hospitalized Patients With ARDS Caused by COVID-19 and H1N1. Chest 2020, 158, 195–205.
- Gattinoni, L.; Chiumello, D.; Caironi, P.; Busana, M.; Romitti, F.; Brazzi, L.; Camporota, L. COVID-19 pneumonia: Different respiratory treatments for different phenotypes? Intensive Care Med. 2020, 46, 1099–1102.
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Int. Med. 2020, 180, 934–943, Erratum in: JAMA Int. Med. 2020, 180, 1031.
- Zhang, P.; Li, J.; Liu, H.; Han, N.; Ju, J.; Kou, Y.; Chen, L.; Jiang, M.; Pan, F.; Zheng, Y.; et al. Correction: Long-term bone and lung consequences associated with hospital-acquired severe acute respiratory syndrome: A 15-year follow-up from a prospective cohort study. Bone Res. 2020, 8, 34, Erratum for: Bone Res. 2020, 8, 8.
- Das, K.M.; Lee, E.Y.; Singh, R.; Enani, M.A.; Al Dossari, K.; Van Gorkom, K.; Larsson, S.G.; Langer, R.D. Follow-up chest radiographic findings in patients with MERS-CoV after recovery. Indian J. Radiol. Imaging 2017, 27, 342–349.
- Karampitsakos, T.; Tzilas, V.; Tringidou, R.; Steiropoulos, P.; Aidinis, V.; Papiris, S.A.; Bouros, D.; Tzouvelekis, A. Lung cancer in patients with idiopathic pulmonary fibrosis. Pulm. Pharmacol. Ther. 2017, 45, 1–10.
- Kato, E.; Takayanagi, N.; Takaku, Y.; Kagiyama, N.; Kanauchi, T.; Ishiguro, T.; Sugita, Y. Incidence and predictive factors of lung cancer in patients with idiopathic pulmonary fibrosis. ERJ Open Res. 2018, 4, 00111–02016.
- Tang, H.; Ren, Y.; She, Y.; Dai, C.; Wang, T.; Su, H.; Sun, W.; Jiang, G.; Chen, C. Is operation safe for lung cancer patients with interstitial lung disease on computed tomography? Ther. Adv. Respir. Dis. 2020, 14, 1753466620971137.
- Luo, Y.H.; Wang, C.; Xu, W.T.; Zhang, Y.; Zhang, T.; Xue, H.; Li, Y.N.; Fu, Z.R.; Wang, Y.; Jin, C.H. 18β-Glycyrrhetinic Acid Has Anti-Cancer Effects via Inducing Apoptosis and G2/M Cell Cycle Arrest, and Inhibiting Migration of A549 Lung Cancer Cells. OncoTargets Ther. 2021, 14, 5131–5144.
- Pallante, P.; Malapelle, U.; Nacchio, M.; Sgariglia, R.; Galati, D.; Capitelli, L.; Zanotta, S.; Galgani, M.; Piemonte, E.; Sanduzzi Zamparelli, A.; et al. Liquid Biopsy Is a Promising Tool for Genetic Testing in Idiopathic Pulmonary Fibrosis. Diagnostics 2021, 11, 1202.
- Sivakumar, P.; Ammar, R.; Thompson, J.R.; Luo, Y.; Streltsov, D.; Porteous, M.; McCoubrey, C.; Cantu, E., 3rd; Beers, M.F.; Jarai, G.; et al. Integrated plasma proteomics and lung transcriptomics reveal novel biomarkers in idiopathic pulmonary fibrosis. Respir. Res. 2021, 22, 273.
- Gomes, R.N.; Manuel, F.; Nascimento, D.S. The bright side of fibroblasts: Molecular signature and regenerative cues in major organs. NPJ Regen. Med. 2021, 6, 43.
- Tan, W.; Wang, Y.; Chen, Y.; Chen, C. Cell tracing reveals the transdifferentiation fate of mouse lung epithelial cells during pulmonary fibrosis in vivo. Exp. Ther. Med. 2021, 22, 1188.
- Liu, X.; Rowan, S.C.; Liang, J.; Yao, C.; Huang, G.; Deng, N.; Xie, T.; Wu, D.; Wang, Y.; Burman, A.; et al. Categorization of lung mesenchymal cells in development and fibrosis. iScience 2021, 24, 102551.
- Wiśniewska, J.; Sadowska, A.; Wójtowicz, A.; Słyszewska, M.; Szóstek-Mioduchowska, A. Perspective on Stem Cell Therapy in Organ Fibrosis: Animal Models and Human Studies. Life 2021, 11, 1068.
- Upagupta, C.; Shimbori, C.; Alsilmi, R.; Kolb, M. Matrix abnormalities in pulmonary fibrosis. Eur. Respir. Rev. 2018, 27, 180033.
- Phan, S.H. Genesis of the myofibroblast in lung injury and fibrosis. Proc. Am. Thorac. Soc. 2012, 9, 148–152.
- Kleaveland, K.R.; Velikoff, M.; Yang, J.; Agarwal, M.; Rippe, R.A.; Moore, B.B.; Kim, K.K. Fibrocytes Are Not an Essential Source of Type I Collagen during Lung Fibrosis. J. Immunol. 2014, 193, 5229–5239.
- Zhou, L.; Yang, K.; Andl, T.; Wickett, R.R.; Zhang, Y. Perspective of Targeting Cancer-Associated Fibroblasts in Melanoma. J. Cancer 2015, 6, 717–726.
- Jena, B.C.; Das, C.K.; Bharadwaj, D.; Mandal, M. Cancer associated fibroblast mediated chemoresistance: A paradigm shift in understanding the mechanism of tumor progression. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188416.
- Joshi, R.S.; Kanugula, S.S.; Sudhir, S.; Pereira, M.P.; Jain, S.; Aghi, M.K. The Role of Cancer-Associated Fibroblasts in Tumor Progression. Cancers 2021, 13, 1399.
- Bochet, L.; Lehuédé, C.; Dauvillier, S.; Wang, Y.Y.; Dirat, B.; Laurent, V.; Dray, C.; Guiet, R.; Maridonneau-Parini, I.; Le Gonidec, S.; et al. Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res. 2013, 73, 5657–5668.
- Raz, Y.; Cohen, N.; Shani, O.; Bell, R.E.; Novitskiy, S.V.; Abramovitz, L.; Levy, C.; Milyavsky, M.; Leider-Trejo, L.; Moses, H.L.; et al. Bone marrow-derived fibroblasts are a functionally distinct stromal cell population in breast cancer. J. Exp. Med. 2018, 215, 3075–3093.
- Petersen, O.W.; Nielsen, H.L.; Gudjonsson, T.; Villadsen, R.; Rønnov-Jessen, L.; Bissell, M.J. The plasticity of human breast carcinoma cells is more than epithelial to mesenchymal conversion. Breast Cancer Res. 2001, 3, 213–217.
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007, 67, 10123–10128.
- Raskov, H. Early mechanisms of tumor dissemination and the relation to surgery. Int. J. Cancer 2020, 3255, 3244–3255.
- Raskov, H.; Orhan, A.; Gaggar, S.; Gögenur, I.; Roz, L. Cancer-Associated Fibroblasts and Tumor-Associated Macrophages in Cancer and Cancer Immunotherapy. Front. Oncol. 2021, 11, 1–17.
- Chilosi, M.; Poletti, V.; Zamò, A.; Lestani, M.; Montagna, L.; Piccoli, P.; Pedron, S.; Bertaso, M.; Scarpa, A.; Murer, B.; et al. Aberrant Wnt/β-catenin pathway activation in idiopathic pulmonary fibrosis. Am. J. Pathol. 2003, 162, 1495–1502.
- Conte, E.; Gili, E.; Fruciano, M.; Korfei, M.; Fagone, E.; Iemmolo, M.; Lo Furno, D.; Giuffrida, R.; Crimi, N.; Guenther, A.; et al. PI3K p110γ overexpression in idiopathic pulmonary fibrosis lung tissue and fibroblast cells: In vitro effects of its inhibition. Lab. Investig. 2013, 93, 566–576.
- Giroux-leprieur, E.; Costantini, A.; Ding, V.W.; He, B. Hedgehog Signaling in Lung Cancer: From Oncogenesis to Cancer Treatment Resistance. Int. J. Mol. Sci. 2018, 19, 2835.
- Wiley, C.D.; Brumwell, A.N.; Davis, S.S.; Jackson, J.R.; Valdovinos, A.; Calhoun, C.; Alimirah, F.; Castellanos, C.A.; Ruan, R.; Wei, Y.; et al. Secretion of leukotrienes by senescent lung fibroblasts promotes pulmonary fibrosis. JCI Insight. 2019, 4, e130056.
- Li, X.; Xie, M.; Lu, C.; Mao, J.; Cao, Y.; Yang, Y.; Wei, Y.; Liu, X.; Cao, S.; Song, Y.; et al. Design and synthesis of Leukotriene A4 hydrolase inhibitors to alleviate idiopathic pulmonary fibrosis and acute lung injury. Eur. J. Med. Chem. 2020, 203, 112614.
- Poczobutt, J.M.; Gijon, M.; Amin, J.; Hanson, D.; Li, H.; Walker, D.; Weiser-Evans, M.; Lu, X.; Murphy, R.C.; Nemenoff, R.A. Eicosanoid profiling in an orthotopic model of lung cancer progression by mass spectrometry demonstrates selective production of leukotrienes by inflammatory cells of the microenvironment. PLoS ONE 2013, 8, e79633.
- Maher, T.M.; Bendstrup, E.; Dron, L.; Langley, J.; Smith, G.; Khalid, J.M.; Patel, H.; Kreuter, M. Global incidence and prevalence of idiopathic pulmonary fibrosis. Respir. Res. 2021, 22, 197.
- Spek, C.A.; Duitman, J. Is idiopathic pulmonary fibrosis a cancer-like disease? Transcriptome analysis to fuel the debate. ERJ Open Res. 2019, 5, 00157–02018.
- JafariNezhad, A.; YektaKooshali, M.H. Lung cancer in idiopathic pulmonary fibrosis: A systematic review and meta-analysis. PLoS ONE 2018, 13, e0202360.
- Ozawa, Y.; Suda, T.; Naito, T.; Enomoto, N.; Hashimoto, D.; Fujisawa, T.; Nakamura, Y.; Inui, N.; Nakamura, H.; Chida, K. Cumulative incidence of and predictive factors for lung cancer in IPF. Respirology 2009, 14, 723–728.
- Tomassetti, S.; Gurioli, C.; Ryu, J.H.; Decker, P.A.; Ravaglia, C.; Tantalocco, P.; Buccioli, M.; Piciucchi, S.; Sverzellati, N.; Dubini, A.; et al. The impact of lung cancer on survival of idiopathic pulmonary fibrosis. Chest 2015, 147, 157–164.
- Alomaish, H.; Ung, Y.; Wang, S.; Tyrrell, P.N.; Zahra, S.A.; Oikonomou, A. Survival analysis in lung cancer patients with interstitial lung disease. PLoS ONE 2021, 16, e0255375.
- d’Alessandro, M.; Bergantini, L.; Torricelli, E.; Cameli, P.; Lavorini, F.; Pieroni, M.; Refini, R.M.; Sestini, P.; Bargagli, E. Systematic Review and Metanalysis of Oncomarkers in IPF Patients and Serial Changes of Oncomarkers in a Prospective Italian Real-Life Case Series. Cancers 2021, 13, 539.
- Goto, T. Measuring Surgery Outcomes of Lung Cancer Patients with Concomitant Pulmonary Fibrosis: A Review of the Literature. Cancers 2018, 10, 223.
- Kawai, T.; Yakumaru, K.; Suzuki, M.; Kageyama, K. Diffuse interstitial pulmonary fibrosis and lung cancer. Acta Pathol. Jpn. 1987, 37, 11–19.
- Hironaka, M.; Fukayama, M. Pulmonary fibrosis and lung carcinoma: A comparative study of metaplastic epithelia in honeycombed areas of usual interstitial pneumonia with or without lung carcinoma. Pathol Int. 1999, 49, 1060–1066.
- Shiratori, T.; Tanaka, H.; Tabe, C.; Tsuchiya, J.; Ishioka, Y.; Itoga, M.; Taima, K.; Takanashi, S.; Tasaka, S. Effect of nintedanib on non-small cell lung cancer in a patient with idiopathic pulmonary fibrosis: A case report and literature review. Thorac. Cancer 2020, 11, 1720–1723.
- Vancheri, C. Common pathways in idiopathic pulmonary fibrosis and cancer. Eur. Respir. Rev. 2013, 22, 265–272.
- Millan-Billi, P.; Serra, C.; Alonso Leon, A.; Castillo, D. Comorbidities, Complications and Non-Pharmacologic Treatment in Idiopathic Pulmonary Fibrosis. Med. Sci. 2018, 6, 59.
- Antoniou, K.M.; Tomassetti, S.; Tsitoura, E.; Vancheri, C. Idiopathic pulmonary fibrosis and lung cancer: A clinical and pathogenesis update. Curr. Opin. Pulm. Med. 2015, 21, 626–633.
- Yokoyama, A.; Kondo, K.; Nakajima, M.; Matsushima, T.; Takahashi, T.; Nishimura, M.; Kohno, N. Prognostic value of circulating KL-6 in idiopathic pulmonary fibrosis. Respirology 2006, 11, 164–168.
- Leuschner, G.; Klotsche, J.; Kreuter, M.; Prasse, A.; Wirtz, H.; Pittrow, D.; Frankenberger, M.; Behr, J.; Kneidinger, N.; INSIGHTS-IPF Registry Group. Idiopathic Pulmonary Fibrosis in Elderly Patients: Analysis of the INSIGHTS-IPF Observational Study. Front. Med. 2020, 7, 601279.
- Cordier, J.F.; Cottin, V. Neglected evidence in idiopathic pulmonary fibrosis: From history to earlier diagnosis. Eur. Respir. J. 2013, 42, 916–923.
- Spagnolo, P.; Ryerson, C.J.; Putman, R.; Oldham, J.; Salisbury, M.; Sverzellati, N.; Valenzuela, C.; Guler, S.; Jones, S.; Wijsenbeek, M.; et al. Early diagnosis of fibrotic interstitial lung disease: Challenges and opportunities. Lancet Respir. Med. 2021, 9, 1065–1076.
- Park, J.; Kim, D.S.; Shim, T.S.; Lim, C.M.; Koh, Y.; Lee, S.D.; Kim, W.S.; Kim, W.D.; Lee, J.S.; Song, K.S. Lung cancer in patients with idiopathic pulmonary fibrosis. Eur. Respir. J. 2001, 17, 1216–1219.
- Wood, D.E.; Kazerooni, E.; Baum, S.L.; Dransfield, M.T.; Eapen, G.A.; Ettinger, D.S.; Hou, L.; Jackman, D.M.; Klippenstein, D.; Kumar, R.; et al. Lung cancer screening, version 1.2015: Featured updates to the NCCN guidelines. J. Natl. Compr. Canc. Netw. 2015, 13, 23–34.
- Humphrey, L.L.; Deffebach, M.; Pappas, M.; Baumann, C.; Artis, K.; Mitchell, J.P.; Zakher, B.; Fu, R.; Slatore, C.G. Screening for lung cancer with low-dose computed tomography: A systematic review to update the US preventive services task force recommendation. Ann. Intern. Med. 2013, 159, 411–420.
- Ley, S.; Ley-Zaporozhan, J. Novelties in imaging in pulmonary fibrosis and nodules. A narrative review. Pulmonology 2020, 26, 39–44.
- Lee, T.; Park, J.Y.; Lee, H.Y.; Cho, Y.J.; Yoon, H.I.; Lee, J.H.; Jheon, S.; Lee, C.T.; Park, J.S. Lung cancer in patients with idiopathic pulmonary fibrosis: Clinical characteristics and impact on survival. Respir. Med. 2014, 108, 1549–1555.
- Goto, T.; Maeshima, A.; Oyamada, Y.; Kato, R. Idiopathic pulmonary fibrosis as a prognostic factor in non-small cell lung cancer. Int. J. Clin. Oncol. 2014, 19, 266–273.
- Spagnolo, P.; Tzouvelekis, A.; Bonella, F. The Management of Patients With Idiopathic Pulmonary Fibrosis. Front. Med. 2018, 5, 148.
- Tzouvelekis, A.; Antoniou, K.; Kreuter, M.; Evison, M.; Blum, T.G.; Poletti, V.; Grigoriu, B.; Vancheri, C.; Spagnolo, P.; Karampitsakos, T.; et al. The DIAMORFOSIS (DIAgnosis and Management Of lung canceR and FibrOSIS) survey: International survey and call for consensus. ERJ Open Res. 2021, 7, 00529–02020.
- Luppi, F.; Kalluri, M.; Faverio, P.; Kreuter, M.; Ferrara, G. Idiopathic pulmonary fibrosis beyond the lung: Understanding disease mechanisms to improve diagnosis and management. Respir. Res. 2021, 22, 109.
- Pastre, J.; Barnett, S.; Ksovreli, I.; Taylor, J.; Brown, A.W.; Shlobin, O.A.; Ahmad, K.; Khangoora, V.; Aryal, S.; King, C.S.; et al. Idiopathic pulmonary fibrosis patients with severe physiologic impairment: Characteristics and outcomes. Respir. Res. 2021, 22, 5.
- Palma, D.A.; Chen, H.; Bahig, H.; Gaede, S.; Harrow, S.; Laba, J.M.; Ryerson, C.J. Assessment of precision irradiation in early non-small cell lung cancer and interstitial lung disease (ASPIRE-ILD): Study protocol for a phase II trial. BMC Cancer 2019, 19, 1206.
- Duitman, J.; van den Ende, T.; Spek, C.A. Immune Checkpoints as Promising Targets for the Treatment of Idiopathic Pulmonary Fibrosis? J. Clin. Med. 2019, 8, 1547.
- Stainer, A.; Faverio, P.; Busnelli, S.; Catalano, M.; Della Zoppa, M.; Marruchella, A.; Pesci, A.; Luppi, F. Molecular Biomarkers in Idiopathic Pulmonary Fibrosis: State of the Art and Future Directions. Int. J. Mol. Sci. 2021, 22, 6255.
- Ni, S.; Song, M.; Guo, W.; Guo, T.; Shen, Q.; Peng, H. Biomarkers and their potential functions in idiopathic pulmonary fibrosis. Expert Rev. Respir. Med. 2020, 14, 593–602.
- Maher, T.M. PROFILEing idiopathic pulmonary fibrosis: Rethinking biomarker discovery. Eur. Respir. Rev. 2013, 22, 148–152.
- Organ, L.A.; Duggan AM, R.; Oballa, E.; Taggart, S.C.; Simpson, J.K.; Kang’ombe, A.R.; Maher, T.M. Biomarkers of collagen synthesis predict progression in the PROFILE idiopathic pulmonary fibrosis cohort. Respir. Res. 2019, 20, 148.
- Peng, D.H.; Luo, Y.; Huang, L.J.; Liao, F.L.; Liu, Y.Y.; Tang, P.; Hu, H.N.; Chen, W. Correlation of Krebs von den Lungen-6 and fibronectin with pulmonary fibrosis in coronavirus disease 2019. Clin. Chim. Acta 2021, 517, 48–53.
- Horimasu, Y.; Hattori, N.; Ishikawa, N.; Kawase, S.; Tanaka, S.; Yoshioka, K.; Costabel, U. Different MUC1 gene polymorphisms in German and Japanese ethnicities affect serum KL-6 levels. Respir. Med. 2012, 106, 1756–1764.
- Korthagen, N.M.; van Moorsel, C.H.; Zanen, P.; Ruven, H.J.; Grutters, J.C. Evaluation of circulating YKL-40 levels in idiopathic interstitial pneumonias. Lung 2014, 192, 975–980.
- Korthagen, N.M.; van Moorsel, C.H.; Barlo, N.P.; Ruven, H.J.; Kruit, A.; Heron, M.; Bosch, J.M.V.D.; Grutters, J.C. Serum and BALF YKL-40 levels are predictors of survival in idiopathic pulmonary fibrosis. Respir. Med. 2011, 105, 106–113.
- Herazo-Maya, J.D.; Sun, J.; Molyneaux, P.L.; Li, Q.; Villalba, J.A.; Tzouvelekis, A.; Lynn, H.; Juan-Guardela, B.M.; Risquez, C.; Osorio, J.C.; et al. Validation of a 52-gene risk profile for outcome prediction in patients with idiopathic pulmonary fibrosis: An international, multicentre, cohort study. Lancet Respir. Med. 2017, 5, 857–868.
- Huang, Y.; Ma, S.-F.; Espindola, M.S.; Vij, R.; Oldham, J.M.; Huffnagle, G.B.; Erb-Downward, J.R.; Flaherty, K.R.; Moore, B.; White, E.S.; et al. Microbes are associated with host innate immune response in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2017, 196, 208–219.
- Molyneaux, P.L.; Willis-Owen, S.A.; Cox, M.J.; James, P.; Cowman, S.; Loebinger, M.; Blanchard, A.; Edwards, L.M.; Stock, C.; Daccord, C.; et al. Host–microbial interactions in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2017, 195, 1640–1650.
- Saini, G.; Jenkins, G.; McKeever, T.; Simpson, J.; Hubbard, R.; Johnson, S.; Braybrooke, R.; Russell, A.-M.; Meakin, G.; Sweeney, L.; et al. The PROFILE study: A prospective study of fibrosis in lung endpoints to discover and qualify biomarkers for use in clinical trials. Am. J. Respir. Crit. Care Med. 2012, 185, A5169.
- Takahashi, H.; Fujishima, T.; Koba, H.; Murakami, S.; Kurokawa, K.; Shibuya, Y.; Shiratori, M.; Kuroki, Y.; Abe, S. Serum surfactant proteins A and D as prognostic factors in idiopathic pulmonary fibrosis and their relationship to disease extent. Am. J. Respir. Crit. Care Med. 2000, 162, 1109–1114.
- Kahn, N.; Rossler, A.K.; Hornemann, K.; Muley, T.; Grünig, E.; Schmidt, W.; Herth, F.J.; Kreuter, M. C-proSP-B: A possible biomarker for pulmonary diseases? Respiration 2018, 96, 117–126.
- Rosas, I.O.; Richards, T.J.; Konishi, K.; Zhang, Y.; Gibson, K.; Lokshin, A.E.; Lindell, K.O.; Cisneros, J.; Macdonald, S.D.; Pardo, A.; et al. MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathic pulmonary fibrosis. PLoS Med. 2008, 5, e93.
- Ebert, W.; Dienemann, H.; Fateh-Moghadam, A.; Scheulen, M.; Konietzko, N.; Schleich, T.; Bombardieri, E. Cytokeratin 19 fragment CYFRA 21-1 compared with carcinoembryonic antigen, squamous cell carcinoma antigen and neuron-specific enolase in lung cancer. Results of an international multicentre study. Eur. J. Clin. Chem. Clin. Biochem. 1994, 32, 189–199.
- Stieber, P.; Hasholzner, U.; Bodenmüller, H.; Nagel, D.; Sunder-Plassmann, L.; Dienemann, H.; Meier, W.; Fateh-Moghadam, A. CYFRA 21-1. A new marker in lung cancer. Cancer 1993, 72, 707–713.
- Albera, C.; Verri, G.; Sciarrone, F.; Sitia, E.; Mangiapia, M.; Solidoro, P. Progressive Fibrosing Interstitial Lung Diseases: A Current Perspective. Biomedicines 2021, 9, 1237.
- Du Bois, R.M.; Weycker, D.; Albera, C.; Bradford, W.Z.; Costabel, U.; Kartashov, A.; King Jr, T.E.; Lancaster, L.; Noble, P.W.; Sahn, S.A.; et al. Forced vital capacity in patients with idiopathic pulmonary fibrosis: Test properties and minimal clinically important difference. Am. J. Respir. Crit. Care Med. 2011, 184, 1382–1389.
- King, T.E., Jr.; Tooze, J.A.; Schwarz, M.I.; Brown, K.R.; Cherniack, R.M. Predicting survival in idiopathic pulmonary fibrosis: Scoring system and survival model. Am. J. Respir. Crit. Care Med. 2001, 164, 1171–1181.
- Lee, S.H.; Park, J.S.; Kim, S.Y.; Kim, D.S.; Kim, Y.W.; Chung, M.P.; Uh, S.T.; Park, C.S.; Park, S.W.; Jeong, S.H.; et al. Comparison of CPI and GAP models in patients with idiopathic pulmonary fibrosis: A nationwide cohort study. Sci. Rep. 2018, 8, 4784.
- Torrisi, S.E.; Ley, B.; Kreuter, M.; Wijsenbeek, M.; Vittinghoff, E.; Collard, H.R.; Vancheri, C. The added value of comorbidities in predicting survival in idiopathic pulmonary fibrosis: A multicentre observational study. Eur. Respir. J. 2019, 53, 1801587.
- Prasse, A.; Probst, C.; Bargagli, E.; Zissel, G.; Toews, G.B.; Flaherty, K.R.; Olschewski, M.; Rottoli, P.; Müller-Quernheim, J. Serum CC-chemokine ligand 18 concentration predicts outcome in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2009, 179, 717–723.
- Richards, T.J.; Kaminski, N.; Baribaud, F.; Flavin, S.; Brodmerkel, C.; Horowitz, D.; Li, K.; Choi, J.; Vuga, L.J.; Lindell, K.O.; et al. Peripheral blood proteins predict mortality in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2012, 185, 67–76.
- Greene, K.E.; Ye, S.; Mason, R.J. Serum surfactant protein-A levels predict development of ARDS in at-risk patients. Chest 1999, 116, 90S–91S.
- Kinder, B.W.; Brown, K.K.; McCormack, F.X.; Ix, J.H.; Kervitsky, A.; Schwarz, M.I.; King, T.E., Jr. Serum surfactant protein-A is a strong predictor of early mortality in idiopathic pulmonary fibrosis. Chest 2009, 135, 1557–1563.
- Nagata, N.; Kitasato, Y.; Wakamatsu, K.; Kawabata, M.; Fukushima, K.; Kajiki, A.; Kitahara, Y.; Watanabe, K. Prognostic value of immunohistochemical surfactant protein A expression in regenerative/hyperplastic alveolar epithelial cells in idiopathic interstitial pneumonias. Diagn. Pathol. 2011, 6, 25.
- Song, J.W.; Song, J.K.; Kim, D.S. Echocardiography and brain natriuretic peptide as prognostic indicators in idiopathic pulmonary fibrosis. Respir. Med. 2009, 103, 180–186.
- Ando, M.; Miyazaki, E.; Ito, T.; Hiroshige, S.; Nureki, S.-I.; Ueno, T.; Takenaka, R.; Fukami, T.; Kumamoto, T. Significance of serum vascular endothelial growth factor level in patients with idiopathic pulmonary fibrosis. Lung 2010, 188, 247–252.
- Gilani, S.R.; Vuga, L.J.; Lindell, K.O.; Gibson, K.F.; Xue, J.; Kaminski, N.; Valentine, V.G.; Lindsay, E.K.; George, M.P.; Steele, C.; et al. CD28 down-regulation on circulating CD4 T-cells is associated with poor prognoses of patients with idiopathic pulmonary fibrosis. PLoS ONE 2010, 5, e8959.
- Kahloon, R.A.; Xue, J.; Bhargava, A.; Csizmadia, E.; Otterbein, L.; Kass, D.J.; Bon, J.; Soejima, M.; Levesque, M.C.; Lindell, K.O.; et al. Patients with idiopathic pulmonary fibrosis with antibodies to heat shock protein 70 have poor prognoses. Am. J. Respir. Crit. Care Med. 2013, 187, 768–775.
- Okamoto, M.; Hoshino, T.; Kitasato, Y.; Sakazaki, Y.; Kawayama, T.; Fujimoto, K.; Ohshima, K.; Shiraishi, H.; Uchida, M.; Ono, J.; et al. Periostin, a matrix protein, is a novel biomarker for idiopathic interstitial pneumonias. Eur. Respir. J. 2011, 37, 1119–1127.
- Tajiri, M.; Okamoto, M.; Fujimoto, K.; Johkoh, T.; Ono, J.; Tominaga, M.; Azuma, K.; Kawayama, T.; Ohta, S.; Izuhara, K.; et al. Serum level of periostin can predict long-term outcome of idiopathic pulmonary fibrosis. Respir. Investig. 2015, 53, 73–81.
- Kadota, J.; Mizunoe, S.; Mito, K.; Mukae, H.; Yoshioka, S.; Kawakami, K.; Koguchi, Y.; Fukushima, K.; Kon, S.; Kohno, S.; et al. High plasma concentrations of osteopontin in patients with interstitial pneumonia. Respir. Med. 2005, 99, 111–117.
- Xue, J.; Kass, D.J.; Bon, J.; Vuga, L.; Tan, J.; Csizmadia, E.; Otterbein, L.; Soejima, M.; Levesque, M.C.; Gibson, K.F.; et al. Plasma B lymphocyte stimulator and B cell differentiation in idiopathic pulmonary fibrosis patients. J. Immunol. 2013, 191, 2089–2095.
- Moeller, A.; Gilpin, S.E.; Ask, K.; Cox, G.; Cook, D.; Gauldie, J.; Margetts, P.J.; Farkas, L.; Dobranowski, J.; Boylan, C.; et al. Circulating fibrocytes are an indicator of poor prognosis in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2009, 179, 588–594.
- Vuga, L.J.; Tedrow, J.R.; Pandit, K.V.; Tan, J.; Kass, D.J.; Xue, J.; Chandra, D.; Leader, J.K.; Gibson, K.F.; Kaminski, N. C-X-C motif chemokine 13 (CXCL13) is a prognostic biomarker of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 189, 966–974.
- Armstrong, H.F.; Podolanczuk, A.J.; Barr, R.G.; Oelsner, E.C.; Kawut, S.M.; Hoffman, E.A.; Tracy, R.; Kaminski, N.; McClelland, R.L.; Lederer, D.J. Serum matrix metalloproteinase-7, respiratory symptoms, and mortality in community-dwelling adults. MESA (Multi-Ethnic Study of Atherosclerosis). Am. J. Respir. Crit. Care Med. 2017, 196, 1311–1317.
- Jenkins, R.G.; Simpson, J.K.; Saini, G.; Bentley, J.H.; Russell, A.M.; Braybrooke, R.; Molyneaux, P.L.; McKeever, T.M.; Wells, A.U.; Flynn, A.; et al. Longitudinal change in collagen degradation biomarkers in idiopathic pulmonary fibrosis: An analysis from the prospective, multicentre PROFILE study. Lancet Respir. Med. 2015, 3, 462–472.
- Maher, T.M.; Braybrooke, R.; Costa, M.J.; Duggan, A.M.; Fahy, W.A.; Hubbard, R.; Lukey, P.; Marshall, R.P.; Oballa, E.; Saini, G.; et al. The PROFILE (Prospective Observation of Fibrosis in the Lung Clinical Endpoints) study. Am. J. Respir. Crit. Care Med. 2017, 195, A6790.
- Maher Toby, M.; Susanne Stowasser Yasuhiko Nishioka Eric, S. Investigating the effects of Nintedanib on biomarkers of extracellular matrix turnover in patients with IPF: Design of the randomised placebo-controlled INMARK® trial. BMJ Open Respir. Res. 2018, 5, e000325.
- Wu, X.; Kim, G.H.; Salisbury, M.L.; Barber, D.; Bartholmai, B.J.; Brown, K.K.; Conoscenti, C.S.; De Backer, J.; Flaherty, K.R.; Gruden, J.F.; et al. Computed tomographic biomarkers in idiopathic pulmonary fibrosis. The future of quantitative analysis. Am. J. Respir. Crit. Care Med. 2019, 199, 12–21.
- Humphries, S.M.; Yagihashi, K.; Huckleberry, J.; Rho, B.-H.; Schroeder, J.D.; Strand, M.; Schwarz, M.I.; Flaherty, K.R.; Kazerooni, E.A.; Van Beek, E.J.R.; et al. Idiopathic pulmonary fibrosis: Data-driven textural analysis of extent of fibrosis at baseline and 15-month follow-up. Radiology 2017, 285, 270–278.
- Jacob, J.; Bartholmai, B.J.; Rajagopalan, S.; Van Moorsel, C.H.M.; Van Es, H.W.; Van Beek, F.T.; Struik, M.H.L.; Kokosi, M.; Egashira, R.; Brun, A.L.; et al. Predicting outcomes in idiopathic pulmonary fibrosis using automated computed tomographic analysis. Am. J. Respir. Crit. Care Med. 2018, 198, 767–776.
- Mammarappallil, J.G.; Rankine, L.; Chan, H.F.; Weatherley, N.; Wild, J.; Driehuys, B. New developments in imaging idiopathic pulmonary fibrosis with hyperpolarized xenon magnetic resonance imaging. J. Thorac. Imaging 2019, 34, 136–150.
- Kwon, B.S.; Choe, J.; Chae, E.J.; Hwang, H.S.; Kim, Y.G.; Song, J.W. Progressive fibrosing interstitial lung disease: Prevalence and clinical outcome. Respir. Res. 2021, 22, 282.
- Soon, P.S.; Kim, E.; Pon, C.K.; Gill, A.J.; Moore, K.; Spillane, A.J.; Benn, D.E.; Baxter, R.C. Breast cancer-associated fibroblasts induce epithelial-to-mesenchymal transition in breast cancer cells. Endocr. Relat. Cancer 2013, 20, 1–12.
- Mathai, S.K.; Schwartz, D.A. Translational research in pulmonary fibrosis. Transl. Res. 2019, 209, 1–13.
- Kekevian, A.; Gershwin, M.E.; Chang, C. Diagnosis and classification of idiopathic pulmonary fibrosis. Autoimmun. Rev. 2014, 13, 508–512.
- Cutting, C.C.; Bowman, W.S.; Dao, N.; Pugashetti, J.V.; Garcia, C.K.; Oldham, J.M.; Newton, C.A. Family History of Pulmonary Fibrosis Predicts Worse Survival in Patients With Interstitial Lung Disease. Chest 2021, 159, 1913–1921.
- Wang, Y.; Kuan, P.J.; Xing, C.; Cronkhite, J.T.; Torres, F.; Rosenblatt, R.L.; DiMaio, J.M.; Kinch, L.N.; Grishin, N.V.; Garcia, C.K. Genetic Defects in Surfactant Protein A2 Are Associated with Pulmonary Fibrosis and Lung Cancer. Am. J. Hum. Genet. 2009, 84, 52–59.
- Song, J.W.; Ogura, T.; Inoue, Y.; Xu, Z.; Quaresma, M.; Stowasser, S.; Stansen, W.; Crestani, B. Long-term treatment with nintedanib in Asian patients with idiopathic pulmonary fibrosis: Results from INPULSIS®-ON. Respirology 2020, 25, 410–416.
- Barratt, S.L.; Mulholland, S.; Al Jbour, K.; Steer, H.; Gutsche, M.; Foley, N.; Srivastava, R.; Sharp, C.; Adamali, H.I. South-West of England’s Experience of the Safety and Tolerability Pirfenidone and Nintedanib for the Treatment of Idiopathic Pulmonary Fibrosis (IPF). Front. Pharmacol. 2018, 9, 1480.
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glassberg, M.K.; Kardatzke, D.; King, T.E., Jr.; Lancaster, L.; Sahn, S.A.; Szwarcberg, J.; et al. Pirfenidone in patients with idiopathic pulmonary fi brosis ( CAPACITY ): Two randomised trials. Lancet 2011, 377, 1760–1769.
- Shin, K.H.; Shin, S.W. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 379, 795–796.
- Feng, H.; Zhao, Y.; Li, Z.; Kang, J. Real-life experiences in a single center: Efficacy of pirfenidone in idiopathic pulmonary fibrosis and fibrotic idiopathic non-specific interstitial pneumonia patients. Ther. Adv. Respir. Dis. 2020, 14, 1753466620963015.
- Allen, R.J.; Porte, J.; Braybrooke, R.; Flores, C.; Fingerlin, T.E.; Oldham, J.M.; Guillen-Guio, B.; Ma, S.F.; Okamoto, T.; John, A.E.; et al. Genetic variants associated with susceptibility to idiopathic pulmonary fibrosis in people of European ancestry: A genome-wide association study. Lancet Respir. Med. 2017, 5, 869–880.
- Allen, R.J.; Guillen-Guio, B.; Oldham, J.M.; Ma, S.F.; Dressen, A.; Paynton, M.L.; Kraven, L.M.; Obeidat, M.; Li, X.; Ng, M.; et al. Genome-wide association study of susceptibility to idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2020, 201, 564–574.
- Shull, J.G.; Pay, M.T.; Lara Compte, C.; Olid, M.; Bermudo, G.; Portillo, K.; Sellarés, J.; Balcells, E.; Vicens-Zygmunt, V.; Planas-Cerezales, L.; et al. Mapping IPF helps identify geographic regions at higher risk for disease development and potential triggers. Respirology 2020, 1–8.
- Padilla, M. Idiopathic pulmonary fibrosis: The role of pathobiology in making a definitive diagnosis. Am. J. Manag. Care 2015, 21 (Suppl. 14), s276–s283.
- Lettieri, S.; Oggionni, T.; Lancia, A.; Bortolotto, C.; Stella, G.M. Immune Stroma in Lung Cancer and Idiopathic Pulmonary Fibrosis: A Common Biologic Landscape? Int. J. Mol. Sci. 2021, 22, 2882.
- Song, M.J.; Kim, S.Y.; Park, M.S.; Kang, M.J.; Lee, S.H.; Park, S.C. A nationwide population-based study of incidence and mortality of lung cancer in idiopathic pulmonary fibrosis. Sci. Rep. 2021, 11, 2596.
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454.
More
Information
Subjects:
Pathology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
24 Nov 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No