Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Virginia Metrangolo | + 1805 word(s) | 1805 | 2021-11-11 12:02:08 | | | |
| 2 | Dean Liu | Meta information modification | 1805 | 2021-11-15 03:16:25 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Metrangolo, V. The Urokinase Receptor in Targeted Cancer Therapy. Encyclopedia. Available online: https://encyclopedia.pub/entry/15971 (accessed on 25 July 2026).
Metrangolo V. The Urokinase Receptor in Targeted Cancer Therapy. Encyclopedia. Available at: https://encyclopedia.pub/entry/15971. Accessed July 25, 2026.
Metrangolo, Virginia. "The Urokinase Receptor in Targeted Cancer Therapy" Encyclopedia, https://encyclopedia.pub/entry/15971 (accessed July 25, 2026).
Metrangolo, V. (2021, November 14). The Urokinase Receptor in Targeted Cancer Therapy. In Encyclopedia. https://encyclopedia.pub/entry/15971
Metrangolo, Virginia. "The Urokinase Receptor in Targeted Cancer Therapy." Encyclopedia. Web. 14 November, 2021.
Copy Citation
The urokinase-type plasminogen activator receptor (uPAR) has now firmly established itself as a versatile molecular target holding promise for the treatment of aggressive malignancies. The copious abundance of uPAR in virtually all human cancerous tissues versus their healthy counterparts has fostered a gradual shift in the therapeutic landscape targeting this receptor from function inhibition to cytotoxic approaches to selectively eradicate the uPAR-expressing cells by delivering a targeted cytotoxic insult.
urokinase plasminogen activator receptor (uPAR)
targeted therapy
cancer treatment
translational research
immunotherapy
tumor stroma
1. Introduction
Since the initial identification, purification, and sequencing of human uPAR were accomplished around 1990 [1][2][3], a still-expanding body of literature documenting uPAR association with cancer has accumulated, and new indications continue to be uncovered. Through the combination of biochemical analysis via site-directed mutagenesis [4][5][6][7] and structural elucidation by crystallography [8][9][10][11][12][13], detailed knowledge about the uPAR structure–function relationships governing the interplay with its two cognate ligands, the serine protease uPA and the provisional matrix protein vitronectin (Vn), was outlined. As the structural and biochemical aspects of uPAR have been extensively investigated and reviewed in detail in [14][5][15][16][17], they are only briefly discussed here and summarized in Figure 1, Figure 2 and Figure 3. The pathophysiological role and expression profile of uPAR shaping its value as a cancer target will be the focus of a more detailed discussion.
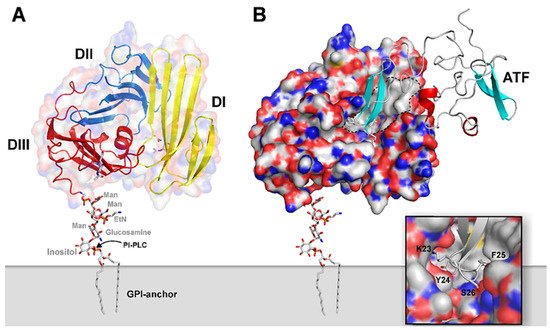
Figure 1. Graphical representation of the structure of uPA•uPAR complexes on the cell surface. A composite molecular model representing human uPAR based on the crystal structure solved for uPAR•ATF complexes is displayed in (A) using the PDB coordinates 2FD6 [8]. The molecular shape of uPAR is visualized by a semitransparent surface, while secondary structure elements are depicted as ribbons. The assembly of the three LU-domains is evident from the color coding, yellow (DI), blue (DII), and red (DIII). A hypothetical model for the GPI-anchor, tethering uPAR to the cell surface, is shown as sticks. In (B), the bimolecular complex of uPAR with its natural binding ligand uPA is illustrated using a solid surface representation for uPAR and a ribbon diagram for the receptor-binding fragment of uPA (ATF) used to crystallize the complex. The large hydrophobic ligand-binding cavity of uPAR is highlighted by the grey area delimited by the hatched black line using the following atomic color coding: grey (C), blue (N), red (O), and yellow (S). The inset in the bottom right corner provides a more detailed illustration of the tight engagement and burial of the tip of the β-hairpin of GFD in uPA within the deepest region of the central cavity in uPAR. Adapted from [18].
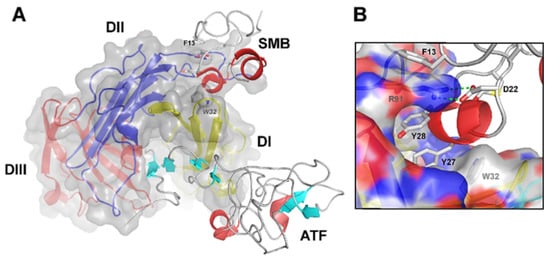
Figure 2. Graphical representation of the crystal structure of the ternary uPA•uPAR•Vn complex. (A) A composite molecular model of the ATF•uPAR•SMB complex solved by X-ray crystallography is shown in (A) using the PDB coordinates 3BTI [9]. The structure is rotated 90° in the horizontal axis compared to Figure 1, providing a “top view” of uPAR and moving the cell surface to the back of the picture. As in Figure 1, uPAR is represented in a composite semitransparent surface and cartoon representation. The bound ligands ATF and SMB (representing uPA and Vn, respectively) are depicted as ribbons. A detailed view of the molecular binding interface between uPAR and SMB in this ternary complex is provided in (B). The corresponding hot spot residues in uPAR (R91 and W32) and SMB (F13, D22, Y27, and Y28) are highlighted and shown as sticks. Adapted from [18].
2. Biological Functions of uPAR
From a historical perspective, uPAR was initially identified as a key regulator of extracellular-matrix (ECM) proteolysis, a fundamental process in the context of cell migration. The high-affinity binding interaction of its bona fide protease ligand uPA is, indeed, instrumental in focalizing plasminogen-activation and subsequent plasmin-mediated proteolytic activity to the cell surface of migrating cells at their leading edge, thereby greatly enhancing the efficiency of the system [19][20][21]. The biochemical aspects of this cascade have been extensively reviewed in [16] and schematically illustrated in Figure 3, which provides a graphical overview of uPAR functional involvement in cancer biology.
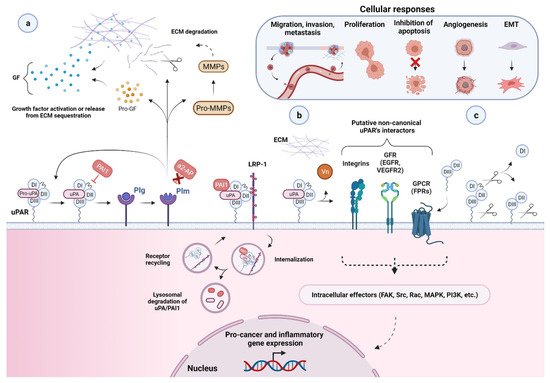
Figure 3. Function and regulation of the urokinase-type plasminogen activator receptor (uPAR) in the biology of cancer. (a) Through a high binding affinity interaction (KD ~0.2 nM), the urokinase receptor binds active uPA and its zymogen pro-uPA, favoring its focused cell-surface activation. Active uPA proteolytically converts the zymogen plasminogen (plg) into active plasmin (plm), which can reciprocally activate pro-uPA, while remaining protected from its primary plasma physiological inhibitors, α2-antiplasmin (α2-AP). These mutual zymogen activation reactions start a powerful positive feedback mechanism, resulting in efficient and localized plasmin generation on the cell surface of migrating cancer cells at their leading edge. The increased cell surface concentration of the reactants involved, respectively, uPA or pro-uPA, by binding to uPAR, and plasmin and plasminogen, via multiple receptors, strongly accelerate and amplify this reciprocal activation loop. Once activated, the broad-spectrum protease plasmin mediates the non-specific proteolysis of several ECM and basement membrane (BM) components, either directly or through the activation of pro-matrix metalloproteinases (pro-MMPs), thereby promoting cancer cell migration, invasion, and metastasis. Plasmin and MMPs can also release or activate ECM-bound cancer-related growth factors (GF) contributing to tumor progression and angiogenesis. Most of these factors then feedback in an autocrine or paracrine fashion to enhance the expression of different pro-cancer genes, including urokinase plasminogen activating system (uPAS) components, such as uPA and uPAR, that further supports the proteolytic cascade and thus tumor progression. Besides α2-AP, another important physiological regulator of uPA-uPAR-induced plg-activation is the serine protease inhibitor (serpin) plasminogen activator inhibitor 1 (PAI1), which specifically inhibits uPA by forming stable ternary complexes with uPAR-bound uPA, which are subsequently internalized via the α2-macroglobulin receptor/low-density lipoprotein receptor-related protein 1 (LRP1). (b) By becoming a part of functional units involving distinct extracellular molecules and membrane co-receptors (e.g., its second main cognate ligand, the matrix protein Vn, members of the integrin adhesion receptor superfamily, G-protein-coupled receptors (GPCR), and growth factors receptors (GFR, e.g., EGFR and VEGFR-2, epidermal growth and vascular endothelial growth factor receptor 2)), uPAR is believed to indirectly choreograph—in a non-proteolytic fashion—several cancer-associated intracellular signal-transduction pathways regulating other tumor hallmarks, including, among others, proliferation, survival, migration, invasion, metastasis, angiogenesis, and epithelial–mesenchymal transition (EMT). The intracellular signaling components are indicated (focal adhesion kinase (FAK), Src, Rac, mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K), etc.), but the pathways remain speculative. A more comprehensive list of uPAR putative non-canonical interacting proteins and related signaling consequences is reported in [22][23][24]. (c) Cell surface uPAR may undergo two major post-translational processing events, namely proteolytic cleavage (in the DI–DII linker region) and shedding (via hydrolysis of the GPI-anchor), resulting in diverse uPAR isoforms, including suPAR D1, suPAR DI–D3, suPAR D2–D3, and GPI-anchored uPAR D2–D3. Created with BioRender.com.
3. uPAR: A Potential “Gateway” for Cytotoxic Cancer Therapy
The pathophysiological role and expression of uPAR in most aggressive cancer lesions, the related prognostic value in many of them, coupled to the apparent lack of overt phenotypes associated with uPAR deficiency, all highlight uPAR as a potential candidate in targeted cancer therapy.
Most experimental strategies explored to date have focused on restraining pericellular uPAR-mediated plasminogen activation, mostly by interfering with the receptor gene expression and interaction with its bona fine ligand, uPA. These include monoclonal antibodies, small molecules- and peptide-derived antagonists (recently reviewed in [25]), recombinant uPA-derived fusion proteins, and various gene therapy approaches. However, although promising in a preclinical setting, none of them have advanced into clinical evaluation. Species-specificity, tumor model limitations, and a rapidly evolving landscape on the relevant determinants and functions of uPAR to target are the main hurdles to the development of uPAR antagonists. The later evidence of uPAR putative involvement in signaling cross-talks with other cancer-associated protein partners has provided an alternative, yet challenging, opportunity to explore therapeutically targeting these interactions and potentially interfere with uPAR functions downstream of uPA proteolytic activity. However, the potential of this approach remains an open question, and future studies elucidating the current controversies underlying uPAR signaling, particularly within in vivo models, will help the field advance.
An in-depth overview of the mentioned approaches, along with the existing challenges hampering their advance into the clinics, is provided by the following detailed reviews [22][26][27][28][29][30][31][23][14][32][33][34][35][16].
The last decade has brought new avenues in cancer treatment focusing on targeted cytotoxic therapies [36]. The widespread overexpression of uPAR in most malignant tissues as compared to their normal counterparts renders uPAR a selective and versatile tool for delivering a direct cytotoxic insult to uPAR expressing cells, leading to their targeted eradication.
This targeting strategy steadily gains momentum and is showing promise in preclinical studies. Different avenues have been and are currently being explored, including immunotherapy approaches. The rationale behind them, related advantages and drawbacks, will be discussed in-depth in the following sections.
In most cases, uPAR targeting is accomplished using monoclonal antibodies, uPA-derived peptides, and a high-affinity receptor-binding fragment of uPA (ATF1-135, which contains the GFD). These ligands provide effective binding scaffolds for conjugation and targeted delivery of different types of cytotoxic payloads or effectors, including traditional anticancer agents, cytotoxic products, radioisotopes, photosensitizers, chimeric antigen receptor (CAR) T-cells, oncolytic virus, or even immunostimulators.
Not only does this approach enhance tumor-specificity, but it also improves intratumoral delivery as uPAR-dependent internalization provides a gateway for targeted intracellular drug release, thereby optimizing the therapeutic response while reducing systemic toxicity. Given the intratumoral heterogeneity in uPAR expression, it is important to characterize this expression pattern when designing an optimal uPAR-targeted cytotoxic insult, as it would strongly impact its effectiveness. The remarkable stromal expression of uPAR also allows targeting this compartment, which may enhance the therapeutic efficacy compared to exclusively targeting the tumor cells, especially in tumors expressing uPAR in both cell types, such as pancreatic cancer, or those lacking a tumor-specific molecular target [27][28][31][37][38][39]. Indeed, while having an indirect anti-cancer effect by attenuating the tumor-promoting effect of the stroma, cytotoxic targeting of this compartment may also increase drug-delivery efficiency by breaking down the dense stromal barrier, which severely hampers tumor perfusion by therapeutic agents, ultimately leading to drug resistance and disease recurrence [40].
In principle, as the tumor stroma predominantly accounts for uPAR expression in most cancer types and patient subgroups, effective tumor regression and/or eradication would ideally be achieved by implementing combined strategies with cytotoxins targeting both the cancer cells per se and the surrounding activated tumor stroma. An intriguing option that is gaining growing interest involves the use of stromal cells, especially tumor-associated macrophages (TAMs) [41][42], as potential autologous delivery vehicles for a localized tumor bystander effect that would indirectly enhance the killing of neighboring cancer cells with low or no uPAR expression. Alternatively, immunosuppressive uPAR-positive TAMs may also be reprogrammed to restore their immunostimulatory/tumoricidal properties and possibly potentiate immune checkpoint blockade therapies (anti-PD-1/PD-L1/CTLA-4 antibodies), as already observed for other therapeutic targets [41][42]. Although still in its infancy, uPAR-mediated stromal targeting is now becoming an attractive avenue, holding promise for the design of combinatorial intervention strategies that may benefit future cancer treatment.
References
- Roldan, A.L.; Cubellis, M.V.; Masucci, M.T.; Behrendt, N.; Lund, L.R.; Danø, K.; Appella, E.; Blasi, F. Cloning and expression of the receptor for human urokinase plasminogen activator, a central molecule in cell surface, plasmin dependent proteolysis. EMBO J. 1990, 9, 467–474.
- Stoppelli, M.P.; Corti, A.; Soffientini, A.; Cassani, G.; Blasi, F.; Assoian, R.K. Differentiation-enhanced binding of the amino-terminal fragment of human urokinase plasminogen activator to a specific receptor on U937 monocytes. Proc. Natl. Acad. Sci. USA 1985, 82, 4939–4943.
- Vassalli, J.D.; Baccino, D.; Belin, D. A cellular binding site for the Mr 55,000 form of the human plasminogen activator, urokinase. J. Cell Biol. 1985, 100, 86–92.
- Gårdsvoll, H.; Gilquin, B.; Le Du, M.H.; Ménèz, A.; Jørgensen, T.J.; Ploug, M. Characterization of the functional epitope on the urokinase receptor. Complete alanine scanning mutagenesis supplemented by chemical cross-linking. J. Biol. Chem. 2006, 281, 19260–19272.
- Gårdsvoll, H.; Ploug, M. Mapping of the vitronectin-binding site on the urokinase receptor: Involvement of a coherent receptor interface consisting of residues from both domain I and the flanking interdomain linker region. J. Biol. Chem. 2007, 282, 13561–13572.
- Gårdsvoll, H.; Danø, K.; Ploug, M. Mapping part of the functional epitope for ligand binding on the receptor for urokinase-type plasminogen activator by site-directed mutagenesis. J. Biol. Chem. 1999, 274, 37995–38003.
- Magdolen, V.; Rettenberger, P.; Koppitz, M.; Goretzki, L.; Kessler, H.; Weidle, U.H.; König, B.; Graeff, H.; Schmitt, M.; Wilhelm, O. Systematic mutational analysis of the receptor-binding region of the human urokinase-type plasminogen activator. Eur. J. Biochem. 1996, 237, 743–751.
- Huai, Q.; Mazar, A.P.; Kuo, A.; Parry, G.C.; Shaw, D.E.; Callahan, J.; Li, Y.; Yuan, C.; Bian, C.; Chen, L.; et al. Structure of human urokinase plasminogen activator in complex with its receptor. Science 2006, 311, 656–659.
- Huai, Q.; Zhou, A.; Lin, L.; Mazar, A.P.; Parry, G.C.; Callahan, J.; Shaw, D.E.; Furie, B.; Furie, B.C.; Huang, M. Crystal structures of two human vitronectin, urokinase and urokinase receptor complexes. Nat. Struct. Mol. Biol. 2008, 15, 422–423.
- Llinas, P.; Le Du, M.H.; Gårdsvoll, H.; Danø, K.; Ploug, M.; Gilquin, B.; Stura, E.A.; Ménez, A. Crystal structure of the human urokinase plasminogen activator receptor bound to an antagonist peptide. EMBO J. 2005, 24, 1655–1663.
- Xu, X.; Gårdsvoll, H.; Yuan, C.; Lin, L.; Ploug, M.; Huang, M. Crystal structure of the urokinase receptor in a ligand-free form. J. Mol. Biol. 2012, 416, 629–641.
- Mertens, H.D.; Kjaergaard, M.; Mysling, S.; Gårdsvoll, H.; Jørgensen, T.J.; Svergun, D.I.; Ploug, M. A flexible multidomain structure drives the function of the urokinase-type plasminogen activator receptor (uPAR). J. Biol. Chem. 2012, 287, 34304–34315.
- Barinka, C.; Parry, G.; Callahan, J.; Shaw, D.E.; Kuo, A.; Bdeir, K.; Cines, D.B.; Mazar, A.; Lubkowski, J. Structural basis of interaction between urokinase-type plasminogen activator and its receptor. J. Mol. Biol. 2006, 363, 482–495.
- Kriegbaum, M.C.; Persson, M.; Haldager, L.; Alpízar-Alpízar, W.; Jacobsen, B.; Gårdsvoll, H.; Kjær, A.; Ploug, M. Rational targeting of the urokinase receptor (uPAR): Development of antagonists and non-invasive imaging probes. Curr. Drug Targets 2011, 12, 1711–1728.
- Kjaergaard, M.; Hansen, L.V.; Jacobsen, B.; Gardsvoll, H.; Ploug, M. Structure and ligand interactions of the urokinase receptor (uPAR). Front. Biosci. 2008, 13, 5441–5461.
- Ploug, M. Structure-function relationships in the interaction between the urokinase-type plasminogen activator and its receptor. Curr. Pharm. Des. 2003, 9, 1499–1528.
- Ploug, M.; Ellis, V. Structure-function relationships in the receptor for urokinase-type plasminogen activator. Comparison to other members of the Ly-6 family and snake venom alpha-neurotoxins. FEBS Lett. 1994, 349, 163–168.
- Ploug, M. Structure-driven design of radionuclide tracers for non-invasive imaging of uPAR and targeted radiotherapy. The tale of a synthetic peptide antagonist. Theranostics 2013, 3, 467–476.
- Ellis, V.; Behrendt, N.; Danø, K. Plasminogen activation by receptor-bound urokinase. A kinetic study with both cell-associated and isolated receptor. J. Biol. Chem. 1991, 266, 12752–12758.
- Ellis, V.; Scully, M.F.; Kakkar, V.V. Plasminogen activation initiated by single-chain urokinase-type plasminogen activator. Potentiation by U937 monocytes. J. Biol. Chem. 1989, 264, 2185–2188.
- Stephens, R.W.; Pöllänen, J.; Tapiovaara, H.; Leung, K.C.; Sim, P.S.; Salonen, E.M.; Rønne, E.; Behrendt, N.; Danø, K.; Vaheri, A. Activation of pro-urokinase and plasminogen on human sarcoma cells: A proteolytic system with surface-bound reactants. J. Cell Biol. 1989, 108, 1987–1995.
- Mahmood, N.; Mihalcioiu, C.; Rabbani, S.A. Multifaceted Role of the Urokinase-Type Plasminogen Activator (uPA) and Its Receptor (uPAR): Diagnostic, Prognostic, and Therapeutic Applications. Front. Oncol. 2018, 8, 24.
- Li Santi, A.; Napolitano, F.; Montuori, N.; Ragno, P. The Urokinase Receptor: A Multifunctional Receptor in Cancer Cell Biology. Therapeutic Implications. Int. J. Mol. Sci. 2021, 22, 4111.
- Eden, G.; Archinti, M.; Furlan, F.; Murphy, R.; Degryse, B. The urokinase receptor interactome. Curr. Pharm. Des. 2011, 17, 1874–1889.
- Yuan, C.; Guo, Z.; Yu, S.; Jiang, L.; Huang, M. Development of inhibitors for uPAR: Blocking the interaction of uPAR with its partners. Drug Discov. Today 2021, 26, 1076–1085.
- Hildenbrand, R.; Niedergethmann, M.; Marx, A.; Belharazem, D.; Allgayer, H.; Schleger, C.; Ströbel, P. Amplification of the urokinase-type plasminogen activator receptor (uPAR) gene in ductal pancreatic carcinomas identifies a clinically high-risk group. Am. J. Pathol. 2009, 174, 2246–2253.
- Lund, I.K.; Illemann, M.; Thurison, T.; Christensen, I.J.; Høyer-Hansen, G. uPAR as anti-cancer target: Evaluation of biomarker potential, histological localization, and antibody-based therapy. Curr. Drug Targets 2011, 12, 1744–1760.
- Mazar, A.P. The urokinase plasminogen activator receptor (uPAR) as a target for the diagnosis and therapy of cancer. Anticancer Drugs 2001, 12, 387–400.
- Montuori, N.; Pesapane, A.; Rossi, F.W.; Giudice, V.; De Paulis, A.; Selleri, C.; Ragno, P. Urokinase type plasminogen activator receptor (uPAR) as a new therapeutic target in cancer. Transl. Med. UniSa 2016, 15, 15–21.
- Ulisse, S.; Baldini, E.; Sorrenti, S.; D’Armiento, M. The urokinase plasminogen activator system: A target for anti-cancer therapy. Curr. Cancer Drug Targets 2009, 9, 32–71.
- Mazar, A.P.; Ahn, R.W.; O’Halloran, T.V. Development of novel therapeutics targeting the urokinase plasminogen activator receptor (uPAR) and their translation toward the clinic. Curr. Pharm. Des. 2011, 17, 1970–1978.
- Ngo, J.C.; Jiang, L.; Lin, Z.; Yuan, C.; Chen, Z.; Zhang, X.; Yu, H.; Wang, J.; Lin, L.; Huang, M. Structural basis for therapeutic intervention of uPA/uPAR system. Curr. Drug Targets 2011, 12, 1729–1743.
- Lin, H.; Xu, L.; Yu, S.; Hong, W.; Huang, M.; Xu, P. Therapeutics targeting the fibrinolytic system. Exp. Mol. Med. 2020, 52, 367–379.
- Pillay, V.; Dass, C.R.; Choong, P.F. The urokinase plasminogen activator receptor as a gene therapy target for cancer. Trends Biotechnol. 2007, 25, 33–39.
- Rockway, T.W.; Nienaber, V.; Giranda, V.L. Inhibitors of the protease domain of urokinase-type plasminogen activator. Curr. Pharm. Des. 2002, 8, 2541–2558.
- Bailly, C. Cell-targeted cytotoxics: A new generation of cytotoxic agents for cancer treatment. Phytochem. Rev. 2014, 13, 171–181.
- Smith, H.W.; Marshall, C.J. Regulation of cell signalling by uPAR. Nat. Rev. Mol. Cell Biol. 2010, 11, 23–36.
- Cantero, D.; Friess, H.; Deflorin, J.; Zimmermann, A.; Bründler, M.A.; Riesle, E.; Korc, M.; Büchler, M.W. Enhanced expression of urokinase plasminogen activator and its receptor in pancreatic carcinoma. Br. J. Cancer 1997, 75, 388–395.
- Bharadwaj, A.G.; Holloway, R.W.; Miller, V.A.; Waisman, D.M. Plasmin and Plasminogen System in the Tumor Microenvironment: Implications for Cancer Diagnosis, Prognosis, and Therapy. Cancers 2021, 13, 1838.
- Valkenburg, K.C.; de Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381.
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904.
- Ngambenjawong, C.; Gustafson, H.H.; Pun, S.H. Progress in tumor-associated macrophage (TAM)-targeted therapeutics. Adv. Drug Deliv. Rev. 2017, 114, 206–221.
More
Information
Subjects:
Oncology; Biotechnology & Applied Microbiology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
876
Revisions:
2 times
(View History)
Update Date:
29 Mar 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No