 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Nikhil Pai | + 4358 word(s) | 4358 | 2021-10-25 11:00:21 | | | |
| 2 | Vivi Li | Meta information modification | 4358 | 2021-11-05 07:39:58 | | |
Video Upload Options
Inflammatory bowel disease (IBD) is a chronic autoimmune condition affecting the gastrointestinal (GI) tract. IBD includes Crohn’s disease (CD) and ulcerative colitis (UC), with UC characterized by inflammation of colonic mucosa and submucosa starting at the rectum and extending through the colon. The precise etiology of UC is unknown but is thought to involve a combination of environmental and genetic factors. Chief among these is the intestinal microbiome, which has been extensively studied both for its role in disease pathogenesis and possible treatment. In this review, we will discuss the microbial changes that have been described in UC, its interplay with host immune function, and evidence supporting its role as a potential therapeutic. We will also discuss parallels between UC, the microbiome and colitis-associated cancer (CAC).
1. Background
2. Microbiome-Immune Interactions in UC
2.1. Immune System Perturbations in UC
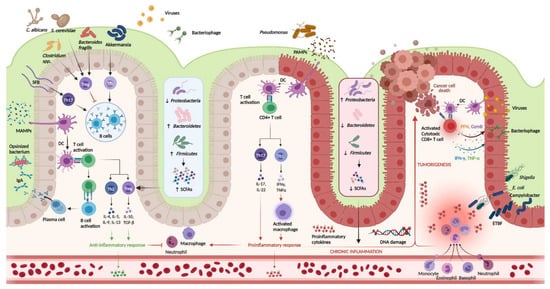
Figure 1. Host-immune interactions in ulcerative colitis. Intestinal microbiota interact with the immune system through various pathways. In the healthy colon, DCs sample MAMPs and present antigens on major histocompatibility complex class II to naive CD4+ T cells. Naive CD4+ T cells become activated and differentiate into various T cell subtypes depending on the presence of specific cytokines within the local microenvironment. Anti-inflammatory Th subtypes comprise Th2 and Treg cells. CD4+ T cells also activate plasma cells which secrete immunoglobulin A (IgA) which is essential for microbial opsonization. Proinflammatory Th subtypes consist of Th1 cells and Th17 cells, which are upregulated in the diseased colon via interactions between DCs and PAMPs. Chronic inflammation contributes to DNA damage and tumorigenesis. Invading viruses stimulate CD8+ cytotoxic T cell activation via antigen-MHC I interactions. However, CD8+ T cells can also assist in cancer cell death. Disruptions in the mucosal barrier provides avenues for microbial translocation, including ETBF, which has been implicated in colitis-associated cancer. Finally, the production of SCFA is increased in the healthy colon (mediated by increased density of Firmicutes and Bacteroidetes phyla), while increased density of the Proteobacterium phylum is associated with lower concentrations of SCFA and colonic inflammation. DC, dendritic cell; DNA, deoxyribonucleic acid; ETBF, enterotoxigenic Bacteroides fragilis; IFN-γ, interferon-gamma; IgA, immunoglobulin A; MAMPs, microbe-associated molecular patterns; PAMPs, pathogen-associated molecular patterns; SCFAs, short chain fatty acids; SFB, segmented filamentous bacteria; Th, T helper; Treg, T regulatory; TNF-α, tumor necrosis factor-alpha. Created in Biorender.com (accessed: 1 August 2021) [9].
2.1.1. Physical Barrier
2.1.2. Immunoglobulin A
2.1.3. Innate and Adaptive Immunity
2.2. Intestinal Microbiota Composition in Ulcerative Colitis
TABLE 1. Intestinal microbiota alterations in ulcerative colitis and impacts on host immune, intestinal function.
| Gut Microbiota Alterations in UC | Consequences for Mammalian Host Health | ||||
|---|---|---|---|---|---|
| Life Domain | Taxonomic Classification | Compositional Changes of Gut Microbiota | Functional Changes of Gut Microbiota | Impact on Host Immune Function |
Impact on Host Intestinal Function |
| Bacteria | Phyla | 16S ribosomal RNA gene sequencing ↓α- diversity in UC as compared with HC [63][71] ↑β-diversity in UC (UC bacteriome clusters differently form HC) [63][71] ↓relative abundance of Firmicutes and Bacteroidetes [13][63][64][67][74] |
shotgun metagenomics sequencing ↑l-arginine biosynthesis (I, IV), biotin biosynthesis II, transfer RNA charging [71] Super pathway of polyamine biosynthesis in patients with risk factors for developing UC as compared with HC [71] ↑amino acid and protein metabolism (in UC as compared with HC): l-lysine fermentation to acetate and butanoate, creatinine degradation II, ketogenesis, protein N-glycosylation [70] ↑proteolytic and elastase activity in pre- and post-UC as compared with HC Correlated with the protease-producing bacterial species altered in UC- Proteobacteria and Bacteroides-↑elastase from B. vulgatus) [71] ↓glycerol and glycerophospholipids in UC as compared with HC Positive correlation between bacterial species and carbohydrate-degradation pathways [75] |
Ruminococcus, Eubacterium, Roseburia, and Akkermansia, Anaerostipes hadrus ↓butyrate production = ↓Treg cells differentiation ↓maturation of Treg cells in the colonic epithelium increased levels of proinflammatory cytokines [64][65][71][72][76] Enterobacteriaceae ↑colonic epithelial cells invasion ↑levels of proinflammatory cytokine IL-8 and TNF-α [77] Fusobacteria ↑tumorigenesis in the colon [65] Faecalibacterium prausnitzii ↑production of IL-12, IFNγ and reduction of IL-10 levels in blood cells [78] Adlercreutzia ↓synthesis of isoflavones, phenolic compounds with antimicrobial and anti-inflammatory properties [71] |
Ruminococcus bromii, Eubacterium rectale, Roseburia, and Akkermansia ↓butyrate production = impaired epithelial barrier function ↑epithelial permeability and commensals translocation [13][64][65] ↑colonic inflammation with crypt abscess [77] ↑of deciduous epithelial and/or blood cells in stools of patients with UC or CAC, gut barrier injury, impaired cell cycle [75] |
| ↑Proteobacteria [13][63][64][65][68] | |||||
| Families | ↓Clostridiaceae [64][65] ↑Enterobacteriaceae [79] |
||||
| Genera | ↓Clostridium clusters IV, XIVa [65] ↓Ruminococcus, Eubacterium, Roseburia, Akkermansia [64][71] ↓Adlercreutzia, Bilophila, Bifidobacterium [71] ↓Bacteroides, Lachnospira, Phascolarctobacterium, Coprococcus, Odoribacter, Butyricimonas [68][79] |
||||
| ↑Escherichia-Shigella, Fusobacterium, Campylobacter, Helicobacter [64][68][71] ↑Actinobacillus [71] ↑Streptococcus, Anaerostipes Enterococcus, Actyinomyces, Lactobacillus, Acetobacter, Rothia, Pseudomonas, Collinsella [68] |
|||||
| Species | ↓Faecalibacterium prausnitzii [65][76][80] ↓Anaerostipes hadrus [72] ↑Flavonifractor plautii, Coprococcus catus, Parabacteroides merdae [71] |
||||
| Fungi | Phyla | Stool ITS2 gene sequencing ↓α-diversity in UC (not in CD) [63] ↑β-diversity between UC in flare as compared with UC in remission and to HC [63] ↑ ratio of Basidiomycota/Ascomycota in UC in flare as compared with UC in remission and to HC [63] ↑correlation between fungi and bacteria in UC as compared with CD and HC [63] Colonic mucosa: ↓fungi load in UC as compared with HC No significant changes in α-diversity UC mycobiota clusters differently from HC No changes in the ratio of Basidiomycota/Ascomycota [81] |
N/A | Saccharomyces cerevisiae and Candida Albicans = ↑IL-6 production [63] ↓Saccharomyces cerevisiae = ↓IL-10 production (anti-inflammatory cytokine) [63] Aspergillus ↑aflatoxin production, a carcinogenic mycotoxin [81] Positive correlation between Wickerhamomyces and Penicillium with the expression of TNF-α and IL-17A, respectively (in colonic mucosa) [81] Negative correlation between Sporobolomyces and IL-6 and between Trametes and IL-1β (in colonic mucosa) [81] |
Aspergillus Potential for aspergillosis, with consequent abdominal pain and GI bleeding [81] |
| Genera | ↓Saccharomyces in UC fecal samples [63] ↑Aspergillus in UC mucosa specimen [81] |
||||
| Species | ↓Saccharomyces cerevisiae in UC fecal samples [63] ↑Candida albicans in UC fecal samples [63] Trend toward an increase in mucosal specimen [82] |
||||
| Virus | Orders | Metagenomics sequencing of viral-like particles ↓α-diversity (virome species richness and evenness) in UC mucosal samples [83] ↑abundance Caudovirales bacteriophages in UC mucosal samples [83] ↑β-diversity; UC mucosal virome clusters differently from HC [83] ↑virome dissimilarity between UC subjects (not observed in HC subjects) [83] |
↓integral component of membrane, DNA binding, ATP-binding cassette (ABC) transporter and integrase core domain in UC as compared with HC [83] ↑Pathways related to the phage lysis of bacteria: DNA template negative regulation of transcription, beta-lactamase, glutamine amidotransferase, glycosal hydrolases, type II/IV secretion system and multicopper oxidase in UC as compared with HC [83] |
↑bacteriophage = ↑bacterial lysis, PAMPs production, TLRs overstimulation, ↑intestinal inflammation [83] ↑transfer of bacterial genetic material (i.e., antibiotic resistance genes) [83] ↑phages can stimulate IFN-γ via the nucleotide-sensing receptor TLR9 [84] |
↑bacteriophages = ↑bacterial lysis, ↑intestinal inflammation, potential implication in abdominal pain, diarrhea [83][84] |
| Families | ↓Anelloviridae (eukaryotic virus) [83] ↑Microviridae (single-stranded DNA phage), Myoviridae, Podoviridae (double-stranded DNA phages) [83] Pneumoviridae (eukaryotic virus) [83] |
||||
| Genera | ↓Coccolithovirus, Minivirus Orthopoxvirus (vertebrate-infecting virus) (all eukaryotic viruses) [83] ↑Phix174microvirus, P1virus, Lambdavirus, T4virus, P22virus (all Caudovirales bacteriophages) Orthopneumovirus [83] |
||||
| Species | ↓α-diversity of Caudovirales species in UC mucosal samples [83] ↑Escherichia and Enterobacteria bacteriophages [83] Lactobacillus, Escherichia, and Bacteroides bacteriophages [84] |
||||
UC, ulcerative colitis; HC, healthy controls; CD, Crohn’s disease; IL, interleukin; CAC, colitis-associated cancer; IFN, interferon; TNF, tumor necrosis factor-α; Treg, regulatory T-cell.
References
- Yamamoto-Furusho, J.K.; Martínez-Benítez, B.; Sánchez-Morales, G.E. Histopathologic Parameters at Diagnosis as Early Predictors of Histologic Remission along the Course of Ulcerative Colitis. Gastroenterol. Res. Pract. 2020, 2020, 1–5.
- Jairath, V.; Feagan, B.G. Global Burden of Inflammatory Bowel Disease. Lancet Gastroenterol. Hepatol. 2020, 5, 2–3.
- Ye, B.D.; McGovern, D.P.B. Genetic Variation in IBD: Progress, Clues to Pathogenesis and Possible Clinical Utility. Expert Rev. Clin. Immunol. 2016, 12, 1091–1107.
- Pigneur, B.; Ruemmele, F.M. Nutritional Interventions for the Treatment of IBD: Current Evidence and Controversies. Ther. Adv. Gastroenterol. 2019, 12, 175628481989053.
- Saidel-Odes, L.; Odes, S. Hygiene Hypothesis in Inflammatory Bowel Disease. Ann. Gastroenterol. 2014, 27, 189–190.
- Troelsen, F.S.; Jick, S. Antibiotic Use in Childhood and Adolescence and Risk of Inflammatory Bowel Disease: A Case-Control Study in the UK Clinical Practice Research Datalink. Inflamm. Bowel Dis. 2020, 26, 440–447.
- Fricke, W.F.; Ravel, J. Microbiome or No Microbiome: Are We Looking at the Prenatal Environment through the Right Lens? Microbiome 2021, 9, 9.
- Wopereis, H.; Oozeer, R.; Knipping, K.; Belzer, C.; Knol, J. The First Thousand Days - Intestinal Microbiology of Early Life: Establishing a Symbiosis. Pediatr. Allergy Immunol. 2014, 25, 428–438.
- BioRender. Available online: www.biorender.com (accessed on 1 August 2021).
- Ananthakrishnan, A.N.; Bernstein, C.N.; Iliopoulos, D.; Macpherson, A.; Neurath, M.F.; Ali, R.A.R.; Vavricka, S.R.; Fiocchi, C. Environmental Triggers in IBD: A Review of Progress and Evidence. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 39–49.
- Agrawal, M.; Sabino, J.; Frias-Gomes, C.; Hillenbrand, C.M.; Soudant, C.; Axelrad, J.E.; Shah, S.C.; Ribeiro-Mourão, F.; Lambin, T.; Peter, I.; et al. Early Life Exposures and the Risk of Inflammatory Bowel Disease: Systematic Review and Meta-Analyses. EClinicalMedicine 2021, 36, 100884.
- Torun, A.; Hupalowska, A.; Trzonkowski, P.; Kierkus, J.; Pyrzynska, B. Intestinal Microbiota in Common Chronic Inflammatory Disorders Affecting Children. Front. Immunol. 2021, 12.
- Pickard, J.M.; Zeng, M.Y.; Caruso, R.; Núñez, G. Gut Microbiota: Role in Pathogen Colonization, Immune Responses, and Inflammatory Disease. Immunol. Rev. 2017, 279, 70–89.
- Belkaid, Y.; Hand, T.W. Role of the Microbiota in Immunity and Inflammation. Cell 2014, 157, 121–141.
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, Stability and Resilience of the Human Gut Microbiota. Nature 2012, 489, 220–230.
- MacPherson, A.J.; Slack, E.; Geuking, M.B.; McCoy, K.D. The Mucosal Firewalls against Commensal Intestinal Microbes. Semin. Immunopathol. 2009, 31, 145–149.
- Johansson, M.E.V.; Holmén Larsson, J.M.; Hansson, G.C. The Two Mucus Layers of Colon Are Organized by the MUC2 Mucin, Whereas the Outer Layer Is a Legislator of Host-Microbial Interactions. Proc. Natl. Acad. Sci. USA 2011, 108, 4659–4665.
- Selma-Royo, M.; Calatayud Arroyo, M.; García-Mantrana, I.; Parra-Llorca, A.; Escuriet, R.; Martínez-Costa, C.; Collado, M.C. Perinatal Environment Shapes Microbiota Colonization and Infant Growth: Impact on Host Response and Intestinal Function. Microbiome 2020, 8.
- Alipour, M.; Zaidi, D.; Valcheva, R.; Jovel, J.; Martínez, I.; Sergi, C.; Walter, J.; Mason, A.L.; Ka-Shu Wong, G.; Dieleman, L.A.; et al. Mucosal Barrier Depletion and Loss of Bacterial Diversity Are Primary Abnormalities in Paediatric Ulcerative Colitis. J. Crohn’s Colitis 2016, 10, 462–471.
- Pothuraju, R.; Krishn, S.R.; Gautam, S.K.; Pai, P.; Ganguly, K.; Chaudhary, S.; Rachagani, S.; Kaur, S.; Batra, S.K. Mechanistic and Functional Shades of Mucins and Associated Glycans in Colon Cancer. Cancers 2020, 12, 649.
- Martens, E.C.; Neumann, M.; Desai, M.S. Interactions of Commensal and Pathogenic Microorganisms with the Intestinal Mucosal Barrier. Nat. Rev. Microbiol. 2018, 16, 457–470.
- Altenbach, D.; Robatzek, S. Pattern Recognition Receptors: From the Cell Surface to Intracellular Dynamics. Mol. Plant-Microbe Interact. 2007, 20, 1031–1039.
- Specian, R.D.; Oliver, M.G. Functional Biology of Intestinal Goblet Cells. Am. J. Physiol.-Cell Physiol. 1991, 260.
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut Biogeography of the Bacterial Microbiota. Nat. Rev. Microbiol. 2015, 14, 20–32.
- McGuckin, M.A.; Lindén, S.K.; Sutton, P.; Florin, T.H. Mucin Dynamics and Enteric Pathogens. Nat. Rev. Microbiol. 2011, 9, 265–278.
- Vanhooren, V.; Vandenbroucke, R.E.; Dewaele, S.; Van Hamme, E.; Haigh, J.J.; Hochepied, T.; Libert, C. Mice Overexpressing β-1,4-Galactosyltransferase i Are Resistant to TNF-Induced Inflammation and DSS-Induced Colitis. PLoS ONE 2013, 8.
- Dorofeyev, A.E.; Vasilenko, I.V.; Rassokhina, O.A.; Kondratiuk, R.B. Mucosal Barrier in Ulcerative Colitis and Crohn’s Disease. Gastroenterol. Res. Pract. 2013, 2013, 431231.
- Yamamoto-Furusho, J.K.; Ascaño-Gutiérrez, I.; Furuzawa-Carballeda, J.; Fonseca-Camarillo, G. Differential Expression of MUC12, MUC16, and MUC20 in Patients with Active and Remission Ulcerative Colitis. Mediat. Inflamm. 2015, 2015, 659018.
- Longman, R.J.; Poulsom, R.; Corfield, A.P.; Warren, B.F.; Wright, N.A.; Thomas, M.G. Alterations in the Composition of the Supramucosal Defense Barrier in Relation to Disease Severity of Ulcerative Colitis. J. Histochem. Cytochem. 2006, 54, 1335–1348.
- Grondin, J.A.; Kwon, Y.H.; Far, P.M.; Haq, S.; Khan, W.I. Mucins in Intestinal Mucosal Defense and Inflammation: Learning From Clinical and Experimental Studies. Front. Immunol. 2020, 11.
- Groschwitz, K.R.; Hogan, S.P. Intestinal Barrier Function: Molecular Regulation and Disease Pathogenesis. J. Allergy Clin. Immunol. 2009, 124, 3–20.
- Vancamelbeke, M.; Vermeire, S. The Intestinal Barrier: A Fundamental Role in Health and Disease. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 821–834.
- Howell, K.J.; Kraiczy, J.; Nayak, K.M.; Gasparetto, M.; Ross, A.; Lee, C.; Mak, T.N.; Koo, B.K.; Kumar, N.; Lawley, T.; et al. DNA Methylation and Transcription Patterns in Intestinal Epithelial Cells from Pediatric Patients with Inflammatory Bowel Diseases Differentiate Disease Subtypes and Associate With Outcome. Gastroenterology 2018, 154, 585–598.
- Kawamoto, A.; Nagata, S.; Anzai, S.; Takahashi, J.; Kawai, M.; Hama, M.; Nogawa, D.; Yamamoto, K.; Kuno, R.; Suzuki, K.; et al. Ubiquitin D Is Upregulated by Synergy of Notch Signalling and TNF-α in the Inflamed Intestinal Epithelia of IBD Patients. J. Crohn’s Colitis 2019, 13, 495–509.
- Blander, J.M. Death in the Intestinal Epithelium—Basic Biology and Implications for Inflammatory Bowel Disease. FEBS J. 2016, 2720–2730.
- Ruder, B.; Atreya, R.; Becker, C. Tumour Necrosis Factor Alpha in Intestinal Homeostasis and Gut Related Diseases. Int. J. Mol. Sci. 2019, 20, 1887.
- Gaujoux, R.; Starosvetsky, E.; Maimon, N.; Vallania, F.; Bar-Yoseph, H.; Pressman, S.; Weisshof, R.; Goren, I.; Rabinowitz, K.; Waterman, M.; et al. Cell-Centred Meta-Analysis Reveals Baseline Predictors of Anti-TNFα Non-Response in Biopsy and Blood of Patients with IBD. Gut 2019, 68, 604–614.
- Brandtzaeg, P.; Johansen, F.E. Mucosal B Cells: Phenotypic Characteristics, Transcriptional Regulation, and Homing Properties. Immunol. Rev. 2005, 206, 32–63.
- Gommerman, J.L.; Rojas, O.L.; Fritz, J.H. Re-Thinking the Functions of IgA+plasma Cells. Gut Microbes 2015, 5, 652–662.
- Pabst, O.; Cerovic, V.; Hornef, M. Secretory IgA in the Coordination of Establishment and Maintenance of the Microbiota. Trends Immunol. 2016, 37, 287–296.
- Hooper, L.V.; Wong, M.H.; Thelin, A.; Hansson, L.; Falk, P.G.; Gordon, J.I. Molecular Analysis of Commensal Host-Microbial Relationships in the Intestine. Science 2001, 291, 881–884.
- Bruno, M.E.C.; Rogier, E.W.; Frantz, A.L.; Stefka, A.T.; Thompson, S.N.; Kaetzel, C.S. Regulation of the Polymeric Immunoglobulin Receptor in Intestinal Epithelial Cells by Enterobacteriaceae: Implications for Mucosal Homeostasis. Immunol. Investig. 2010, 39, 356–382.
- Wei, H.; Wang, J.Y. Role of Polymeric Immunoglobulin Receptor in Iga and Igm Transcytosis. Int. J. Mol. Sci. 2021, 22, 2284.
- Johansen, F.E.; Kaetzel, C.S. Regulation of the Polymeric Immunoglobulin Receptor and IgA Transport: New Advances in Environmental Factors That Stimulate PIgR Expression and Its Role in Mucosal Immunity. Mucosal Immunol. 2011, 4, 598–602.
- Van der Steen, L.; Tuk, C.W.; Bakema, J.E.; Kooij, G.; Reijerkerk, A.; Vidarsson, G.; Bouma, G.; Kraal, G.; de Vries, H.E.; Beelen, R.H.J.; et al. Immunoglobulin A: FcαRI Interactions Induce Neutrophil Migration Through Release of Leukotriene B4. Gastroenterology 2009, 137.
- Traicoff, J.L.; De Marchis, L.; Ginsburg, B.L.; Zamora, R.E.; Khattar, N.H.; Blanch, V.J.; Plummer, S.; Bargo, S.A.; Templeton, D.J.; Casey, G.; et al. Characterization of the Human Polymeric Immunoglobulin Receptor (PIGR) 3′UTR and Differential Expression of PIGR MRNA during Colon Tumorigenesis. J. Biomed. Sci. 2003, 10, 792–804.
- Hurtado, C.G.; Wan, F.; Housseau, F.; Sears, C.L. Roles for Interleukin 17 and Adaptive Immunity in Pathogenesis of Colorectal Cancer. Gastroenterology 2018, 155, 1706–1715.
- Caputi, V.; Popov, J.; Giron, M.C.; O’Mahony, S. Gut Microbiota as a Mediator of Host Neuro-Immune Interactions: Implications in Neuroinflammatory Disorders. Mod. Trends Psychiatry 2021, 32, 40–57.
- Rescigno, M. CCR6+ Dendritic Cells: The Gut Tactical-Response Unit. Immunity 2006, 24, 508–510.
- Sun, T.; Nguyen, A.; Gommerman, J.L. Dendritic Cell Subsets in Intestinal Immunity and Inflammation. J. Immunol. 2020, 204, 1075–1083.
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Philip Schumm, L.; Sharma, Y.; Anderson, C.A.; et al. Host-Microbe Interactions Have Shaped the Genetic Architecture of Inflammatory Bowel Disease. Nature 2012, 491, 119–124.
- Steinbach, E.C.; Plevy, S.E. The Role of Macrophages and Dendritic Cells in the Initiation of Inflammation in IBD. Inflamm. Bowel Dis. 2014, 20, 166–175.
- Hart, A.L.; Al-Hassi, H.O.; Rigby, R.J.; Bell, S.J.; Emmanuel, A.V.; Knight, S.C.; Kamm, M.A.; Stagg, A.J. Characteristics of Intestinal Dendritic Cells in Inflammatory Bowel Diseases. Gastroenterology 2005, 129, 50–65.
- Verstockt, B.; Ferrante, M.; Vermeire, S.; Van Assche, G. New Treatment Options for Inflammatory Bowel Diseases. J. Gastroenterol. 2018, 53, 585–590.
- Heller, F.; Fromm, A.; Gitter, A.H.; Mankertz, J.; Schulzke, J.D. Epithelial Apoptosis Is a Prominent Feature of the Epithelial Barrier Disturbance in Intestinal Inflammation: Effect of pro-Inflammatory Interleukin-13 on Epithelial Cell Function. Mucosal Immunol. 2008, 1, 58–61.
- Tan, T.G.; Sefik, E.; Geva-Zatorsky, N.; Kua, L.; Naskar, D.; Teng, F.; Pasman, L.; Ortiz-Lopez, A.; Jupp, R.; Wu, H.J.J.; et al. Identifying Species of Symbiont Bacteria from the Human Gut That, Alone, Can Induce Intestinal Th17 Cells in Mice. Proc. Natl. Acad. Sci. USA 2016, 113, E8141–E8150.
- Erturk-Hasdemir, D.; Oh, S.F.; Okan, N.A.; Stefanetti, G.; Gazzaniga, F.S.; Seeberger, P.H.; Plevy, S.E.; Kasper, D.L. Symbionts Exploit Complex Signaling to Educate the Immune System. Proc. Natl. Acad. Sci. USA 2019, 116, 26157–26166.
- Zhao, F.; Qu, J.; Wang, W.; Li, S.; Xu, S. The Imbalance of Th1/Th2 Triggers an Inflammatory Response in Chicken Spleens after Ammonia Exposure. Poult. Sci. 2020, 99, 3817–3822.
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14.
- Ananthakrishnan, A.N. Epidemiology and Risk Factors for IBD. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 205–217.
- Lyte, J.M. Eating for 3.8 × 1013: Examining the Impact of Diet and Nutrition on the Microbiota-Gut-Brain Axis through the Lens of Microbial Endocrinology. Front. Endocrinol. 2019, 10.
- Pei, L.Y.; Ke, Y.S.; Zhao, H.H.; Wang, L.; Jia, C.; Liu, W.Z.; Fu, Q.H.; Shi, M.N.; Cui, J.; Li, S.C. Role of Colonic Microbiota in the Pathogenesis of Ulcerative Colitis. BMC Gastroenterol. 2019, 19.
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal Microbiota Dysbiosis in IBD. Gut 2017, 66, 1039–1048.
- Zou, J.; Liu, C.; Jiang, S.; Qian, D.; Duan, J. Cross Talk between Gut Microbiota and Intestinal Mucosal Immunity in the Development of Ulcerative Colitis. Infect. Immun. 2021, 89.
- Pavel, F.M.; Vesa, C.M.; Gheorghe, G.; Diaconu, C.C.; Stoicescu, M.; Munteanu, M.A.; Babes, E.E.; Tit, D.M.; Toma, M.M.; Bungau, S. Highlighting the Relevance of Gut Microbiota Manipulation in Inflammatory Bowel Disease. Diagnostics 2021, 11, 1090.
- Frank, D.N.; St., Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-Phylogenetic Characterization of Microbial Community Imbalances in Human Inflammatory Bowel Diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785.
- Chen, D.L.; Dai, Y.C.; Zheng, L.; Chen, Y.L.; Zhang, Y.L.; Tang, Z.P. Features of the Gut Microbiota in Ulcerative Colitis Patients with Depression: A Pilot Study. Medicine 2021, 100, e24845.
- Xu, N.; Bai, X.; Cao, X.; Yue, W.; Jiang, W.; Yu, Z. Changes in Intestinal Microbiota and Correlation with TLRs in Ulcerative Colitis in the Coastal Area of Northern China. Microb. Pathog. 2021, 150.
- Fernandes, P.; Macsharry, J.; Darby, T.; Fanning, A.; Shanahan, F.; Houston, A.; Brint, E. Differential Expression of Key Regulators of Toll-like Receptors in Ulcerative Colitis and Crohn’s Disease: A Role for Tollip and Peroxisome Proliferator-Activated Receptor Gamma? Clin. Exp. Immunol. 2016, 183, 358–368.
- Franchimont, D.; Vermeire, S.; El Housni, H.; Pierik, M.; Van Steen, K.; Gustot, T.; Quertinmont, E.; Abramowicz, M.; Van Gossum, A.; Devière, J.; et al. Deficient Host-Bacteria Interactions in Inflammatory Bowel Disease? The Toll-like Receptor (TLR)-4 Asp299gly Polymorphism Is Associated with Crohn’s Disease and Ulcerative Colitis. Gut 2004, 53, 987–992.
- Galipeau, H.J.; Caminero, A.; Turpin, W.; Bermudez-Brito, M.; Santiago, A.; Libertucci, J.; Constante, M.; Raygoza Garay, J.A.; Rueda, G.; Armstrong, S.; et al. Novel Fecal Biomarkers That Precede Clinical Diagnosis of Ulcerative Colitis. Gastroenterology 2021, 160, 1532–1545.
- Wiechers, C.; Zou, M.; Galvez, E.; Beckstette, M.; Ebel, M.; Strowig, T.; Huehn, J.; Pezoldt, J. The Microbiota Is Dispensable for the Early Stages of Peripheral Regulatory T Cell Induction within Mesenteric Lymph Nodes. Cell. Mol. Immunol. 2021, 18, 1211–1221.
- Atarashi, K.; Tanoue, T.; Oshima, K.; Suda, W.; Nagano, Y.; Nishikawa, H.; Fukuda, S.; Saito, T.; Narushima, S.; Hase, K.; et al. Treg Induction by a Rationally Selected Mixture of Clostridia Strains from the Human Microbiota. Nature 2013, 500, 232–236.
- Machiels, K.; Joossens, M.; Sabino, J.; De Preter, V.; Arijs, I.; Eeckhaut, V.; Ballet, V.; Claes, K.; Van Immerseel, F.; Verbeke, K.; et al. A Decrease of the Butyrate-Producing Species Roseburia Hominis and Faecalibacterium Prausnitzii Defines Dysbiosis in Patients with Ulcerative Colitis. Gut 2014, 63, 1275–1283.
- Jiang, P.; Wu, S.; Luo, Q.; Zhao, X.; Chen, W.-H. Metagenomic Analysis of Common Intestinal Diseases Reveals Relationships among Microbial Signatures and Powers Multidisease Diagnostic Models. mSystems 2021, 6.
- Ryan, F.J.; Ahern, A.M.; Fitzgerald, R.S.; Laserna-Mendieta, E.J.; Power, E.M.; Clooney, A.G.; O’Donoghue, K.W.; McMurdie, P.J.; Iwai, S.; Crits-Christoph, A.; et al. Colonic Microbiota Is Associated with Inflammation and Host Epigenomic Alterations in Inflammatory Bowel Disease. Nat. Commun. 2020, 11.
- Ohkusa, T.; Yoshida, T.; Sato, N.; Watanabe, S.; Tajiri, H.; Okayasu, I. Commensal Bacteria Can Enter Colonic Epithelial Cells and Induce Proinflammatory Cytokine Secretion: A Possible Pathogenic Mechanism of Ulcerative Colitis. J. Med. Microbiol. 2009, 58, 535–545.
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermúdez-Humarán, L.G.; Gratadoux, J.-J.J.J.; Blugeon, S.; Bridonneau, C.; Furet, J.P.J.-P.; Corthier, G.; et al. Faecalibacterium Prausnitzii Is an Anti-Inflammatory Commensal Bacterium Identified by Gut Microbiota Analysis of Crohn Disease Patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736.
- Nishihara, Y.; Ogino, H.; Tanaka, M.; Ihara, E.; Fukaura, K.; Nishioka, K.; Chinen, T.; Tanaka, Y.; Nakayama, J.; Kang, D.; et al. Mucosa-Associated Gut Microbiota Reflects Clinical Course of Ulcerative Colitis. Sci. Rep. 2021, 11.
- Kedia, S.; Ghosh, T.S.; Jain, S.; Desigamani, A.; Kumar, A.; Gupta, V.; Bopanna, S.; Yadav, D.P.; Goyal, S.; Makharia, G.; et al. Gut Microbiome Diversity in Acute Severe Colitis Is Distinct from Mild to Moderate Ulcerative Colitis. J. Gastroenterol. Hepatol. 2021, 36, 731–739.
- Qiu, X.; Ma, J.; Jiao, C.; Mao, X.; Zhao, X.; Lu, M.; Wang, K.; Zhang, H. Alterations in the Mucosa-Associated Fungal Microbiota in Patients with Ulcerative Colitis. Oncotarget 2017, 8, 107577–107588.
- Hoarau, G.; Mukherjee, P.K.; Gower-Rousseau, C.; Hager, C.; Chandra, J.; Retuerto, M.A.; Neut, C.; Vermeire, S.; Clemente, J.; Colombel, J.F.; et al. Bacteriome and Mycobiome Interactions Underscore Microbial Dysbiosis in Familial Crohn’s Disease. MBio 2016, 7.
- Zuo, T.; Lu, X.J.; Zhang, Y.; Cheung, C.P.; Lam, S.; Zhang, F.; Tang, W.; Ching, J.Y.L.; Zhao, R.; Chan, P.K.S.; et al. Gut Mucosal Virome Alterations in Ulcerative Colitis. Gut 2019, 68, 1169–1179.
- Gogokhia, L.; Buhrke, K.; Bell, R.; Hoffman, B.; Brown, D.G.; Hanke-Gogokhia, C.; Ajami, N.J.; Wong, M.C.; Ghazaryan, A.; Valentine, J.F.; et al. Expansion of Bacteriophages Is Linked to Aggravated Intestinal Inflammation and Colitis. Cell Host Microbe 2019, 25, 285–299.e8.
- Friswell, M.; Campbell, B.; Rhodes, J. The Role of Bacteria in the Pathogenesis of Inflammatory Bowel Disease. Gut Liver 2010, 4, 295–306.
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-Specific Alterations in the Enteric Virome in Inflammatory Bowel Disease. Cell 2015, 160, 447–460.
- Lavelle, A.; Sokol, H. Gut Microbiota-Derived Metabolites as Key Actors in Inflammatory Bowel Disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 223–237.
- Sinopoulou, V.; Gordon, M.; Dovey, T.M.; Akobeng, A.K. Interventions for the Management of Abdominal Pain in Ulcerative Colitis. Cochrane Database Syst. Rev. 2021, 2021.

