 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Xiaogang Li | + 2391 word(s) | 2391 | 2021-10-25 12:19:51 | | | |
| 2 | Jessie Wu | Meta information modification | 2391 | 2021-10-26 06:13:22 | | | | |
| 3 | Jessie Wu | Meta information modification | 2391 | 2021-11-01 04:54:51 | | |
Video Upload Options
Mitochondria are heterogeneous and highly dynamic organelles, playing critical roles in adenosine triphosphate (ATP) synthesis, metabolic modulation, reactive oxygen species (ROS) generation, and cell differentiation and death. Mitochondrial dysfunction has been recognized as a contributor in many diseases. The kidney is an organ enriched in mitochondria and with high energy demand in the human body. Recent studies have been focusing on how mitochondrial dysfunction contributes to the pathogenesis of different forms of kidney diseases, including acute kidney injury (AKI) and chronic kidney disease (CKD). AKI has been linked to an increased risk of developing CKD. AKI and CKD have a broad clinical syndrome and a substantial impact on morbidity and mortality, encompassing various etiologies and representing important challenges for global public health. Renal mitochondrial disorders are a common feature of diverse forms of AKI and CKD, which result from defects in mitochondrial structure, dynamics, and biogenesis as well as crosstalk of mitochondria with other organelles. Persistent dysregulation of mitochondrial homeostasis in AKI and CKD affects diverse cellular pathways, leading to an increase in renal microvascular loss, oxidative stress, apoptosis, and eventually renal failure. It is important to understand the cellular and molecular events that govern mitochondria functions and pathophysiology in AKI and CKD, which should facilitate the development of novel therapeutic strategies. This review provides an overview of the molecular insights of the mitochondria and the specific pathogenic mechanisms of mitochondrial dysfunction in the progression of AKI, CKD, and AKI to CKD transition. We also discuss the possible beneficial effects of mitochondrial-targeted therapeutic agents for the treatment of mitochondrial dysfunction-mediated AKI and CKD, which may translate into therapeutic options to ameliorate renal injury and delay the progression of these kidney diseases.
1. The Roles of Mitochondrial in Acute kidney injury, Chronic kidney disease , and Acute kidney injury to Chronic kidney disease Transition
2. The Roles of Mitochondrial Structure, Dynamics, and Biogenesis in AKI
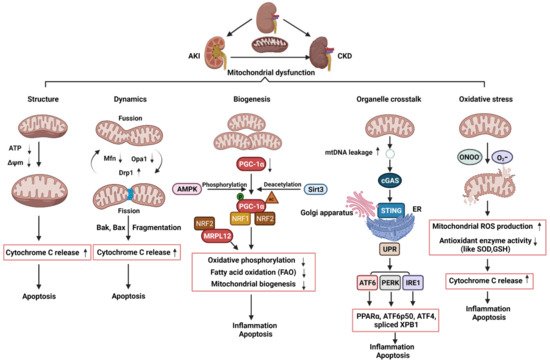
3. The Roles of Mitochondrial Dynamics and Biogenesis in CKD
4. The Roles of Mitochondrial Dysfunction and Its Crosstalk with ER in the Transition of AKI to CKD
References
- Jiang, M.; Bai, M.; Lei, J.; Xie, Y.; Xu, S.; Jia, Z.; Zhang, A. Mitochondrial dysfunction and the AKI-to-CKD transition. Am. J. Physiol. Ren. Physiol. 2020, 319, F1105–F1116.
- Lan, R.; Geng, H.; Singha, P.K.; Saikumar, P.; Bottinger, E.P.; Weinberg, J.M.; Venkatachalam, M.A. Mitochondrial Pathology and Glycolytic Shift during Proximal Tubule Atrophy after Ischemic AKI. J. Am. Soc. Nephrol. 2016, 27, 3356–3367.
- Wakabayashi, T.; Karbowski, M. Structural changes of mitochondria related to apoptosis. Biol. Signals Recept. 2001, 10, 26–56.
- Guan, N.; Ren, Y.L.; Liu, X.Y.; Zhang, Y.; Pei, P.; Zhu, S.N.; Fan, Q. Protective role of cyclosporine A and minocycline on mitochondrial disequilibrium-related podocyte injury and proteinuria occurrence induced by adriamycin. Nephrol. Dial. Transpl. 2015, 30, 957–969.
- Yan, Y.; Ma, Z.; Zhu, J.; Zeng, M.; Liu, H.; Dong, Z. miR-214 represses mitofusin-2 to promote renal tubular apoptosis in ischemic acute kidney injury. Am. J. Physiol. Ren. Physiol. 2020, 318, F878–F887.
- Brooks, C.; Wei, Q.; Cho, S.G.; Dong, Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J. Clin. Investig. 2009, 119, 1275–1285.
- Brooks, C.; Cho, S.G.; Wang, C.Y.; Yang, T.; Dong, Z. Fragmented mitochondria are sensitized to Bax insertion and activation during apoptosis. Am. J. Physiol. Cell Physiol. 2011, 300, C447–C455.
- Saikumar, P.; Dong, Z.; Patel, Y.; Hall, K.; Hopfer, U.; Weinberg, J.M.; Venkatachalam, M.A. Role of hypoxia-induced Bax translocation and cytochrome c release in reoxygenation injury. Oncogene 1998, 17, 3401–3415.
- Mikhailov, V.; Mikhailova, M.; Degenhardt, K.; Venkatachalam, M.A.; White, E.; Saikumar, P. Association of Bax and Bak homo-oligomers in mitochondria. Bax requirement for Bak reorganization and cytochrome c release. J. Biol. Chem. 2003, 278, 5367–5376.
- Tanaka, T.; Nangaku, M.; Miyata, T.; Inagi, R.; Ohse, T.; Ingelfinger, J.R.; Fujita, T. Blockade of calcium influx through L-type calcium channels attenuates mitochondrial injury and apoptosis in hypoxic renal tubular cells. J. Am. Soc. Nephrol. 2004, 15, 2320–2333.
- Wei, Q.; Dong, G.; Chen, J.K.; Ramesh, G.; Dong, Z. Bax and Bak have critical roles in ischemic acute kidney injury in global and proximal tubule-specific knockout mouse models. Kidney Int. 2013, 84, 138–148.
- Xiao, X.; Hu, Y.; Quiros, P.M.; Wei, Q.; Lopez-Otin, C.; Dong, Z. OMA1 mediates OPA1 proteolysis and mitochondrial fragmentation in experimental models of ischemic kidney injury. Am. J. Physiol. Ren. Physiol. 2014, 306, F1318–F1326.
- Perry, H.M.; Huang, L.; Wilson, R.J.; Bajwa, A.; Sesaki, H.; Yan, Z.; Rosin, D.L.; Kashatus, D.F.; Okusa, M.D. Dynamin-Related Protein 1 Deficiency Promotes Recovery from AKI. J. Am. Soc. Nephrol. 2018, 29, 194–206.
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1alpha Signaling Pathways. Front. Genet. 2019, 10, 435.
- Lynch, M.R.; Tran, M.T.; Parikh, S.M. PGC1alpha in the kidney. Am. J. Physiol. Ren. Physiol. 2018, 314, F1–F8.
- Tran, M.T.; Zsengeller, Z.K.; Berg, A.H.; Khankin, E.V.; Bhasin, M.K.; Kim, W.; Clish, C.B.; Stillman, I.E.; Karumanchi, S.A.; Rhee, E.P.; et al. PGC1alpha drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 2016, 531, 528–532.
- Portilla, D.; Dai, G.; McClure, T.; Bates, L.; Kurten, R.; Megyesi, J.; Price, P.; Li, S. Alterations of PPARalpha and its coactivator PGC-1 in cisplatin-induced acute renal failure. Kidney Int. 2002, 62, 1208–1218.
- Morigi, M.; Perico, L.; Rota, C.; Longaretti, L.; Conti, S.; Rottoli, D.; Novelli, R.; Remuzzi, G.; Benigni, A. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Investig. 2015, 125, 715–726.
- Tran, M.; Tam, D.; Bardia, A.; Bhasin, M.; Rowe, G.C.; Kher, A.; Zsengeller, Z.K.; Akhavan-Sharif, M.R.; Khankin, E.V.; Saintgeniez, M.; et al. PGC-1alpha promotes recovery after acute kidney injury during systemic inflammation in mice. J. Clin. Investig. 2011, 121, 4003–4014.
- Parikh, S.M.; Yang, Y.; He, L.; Tang, C.; Zhan, M.; Dong, Z. Mitochondrial function and disturbances in the septic kidney. Semin. Nephrol. 2015, 35, 108–119.
- Jang, H.S.; Noh, M.R.; Kim, J.; Padanilam, B.J. Defective Mitochondrial Fatty Acid Oxidation and Lipotoxicity in Kidney Diseases. Front. Med. (Lausanne) 2020, 7, 65.
- Galvan, D.L.; Green, N.H.; Danesh, F.R. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int. 2017, 92, 1051–1057.
- Xiao, L.; Zhu, X.; Yang, S.; Liu, F.; Zhou, Z.; Zhan, M.; Xie, P.; Zhang, D.; Li, J.; Song, P.; et al. Rap1 ameliorates renal tubular injury in diabetic nephropathy. Diabetes 2014, 63, 1366–1380.
- Zhan, M.; Usman, I.M.; Sun, L.; Kanwar, Y.S. Disruption of renal tubular mitochondrial quality control by Myo-inositol oxygenase in diabetic kidney disease. J. Am. Soc. Nephrol. 2015, 26, 1304–1321.
- Galvan, D.L.; Long, J.; Green, N.; Chang, B.H.; Lin, J.S.; Schumacker, P.; Truong, L.D. Drp1S600 phosphorylation regulates mitochondrial fission and progression of nephropathy in diabetic mice. J. Clin. Investig. 2019, 129, 2807–2823.
- Ayanga, B.A.; Badal, S.S.; Wang, Y.; Galvan, D.L.; Chang, B.H.; Schumacker, P.T.; Danesh, F.R. Dynamin-Related Protein 1 Deficiency Improves Mitochondrial Fitness and Protects against Progression of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2016, 27, 2733–2747.
- Qin, X.; Zhao, Y.; Gong, J.; Huang, W.; Su, H.; Yuan, F.; Fang, K.; Wang, D.; Li, J.; Zou, X.; et al. Berberine Protects Glomerular Podocytes via Inhibiting Drp1-Mediated Mitochondrial Fission and Dysfunction. Theranostics 2019, 9, 1698–1713.
- Wang, Y.; Lu, M.; Xiong, L.; Fan, J.; Zhou, Y.; Li, H.; Peng, X.; Zhong, Z.; Wang, Y.; Huang, F.; et al. Drp1-mediated mitochondrial fission promotes renal fibroblast activation and fibrogenesis. Cell Death Dis. 2020, 11, 29.
- Platt, C.; Coward, R.J. Peroxisome proliferator activating receptor-gamma and the podocyte. Nephrol. Dial. Transpl. 2017, 32, 423–433.
- Yuan, Y.; Huang, S.; Wang, W.; Wang, Y.; Zhang, P.; Zhu, C.; Ding, G.; Liu, B.; Yang, T.; Zhang, A. Activation of peroxisome proliferator-activated receptor-gamma coactivator 1alpha ameliorates mitochondrial dysfunction and protects podocytes from aldosterone-induced injury. Kidney Int. 2012, 82, 771–789.
- Ishimoto, Y.; Inagi, R.; Yoshihara, D.; Kugita, M.; Nagao, S.; Shimizu, A.; Takeda, N.; Wake, M.; Honda, K.; Zhou, J.; et al. Mitochondrial Abnormality Facilitates Cyst Formation in Autosomal Dominant Polycystic Kidney Disease. Mol. Cell Biol. 2017, 37, e00337-17.
- Sharma, K.; Karl, B.; Mathew, A.V.; Gangoiti, J.A.; Wassel, C.L.; Saito, R.; Pu, M.; Sharma, S.; You, Y.H.; Wang, L.; et al. Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. J. Am. Soc. Nephrol. 2013, 24, 1901–1912.
- Han, S.H.; Wu, M.Y.; Nam, B.Y.; Park, J.T.; Yoo, T.H.; Kang, S.W.; Park, J.; Chinga, F.; Li, S.Y.; Susztak, K. PGC-1alpha Protects from Notch-Induced Kidney Fibrosis Development. J. Am. Soc. Nephrol. 2017, 28, 3312–3322.
- Brinkkoetter, P.T.; Bork, T.; Salou, S.; Liang, W.; Mizi, A.; Ozel, C.; Koehler, S.; Hagmann, H.H.; Ising, C.; Kuczkowski, A.; et al. Anaerobic Glycolysis Maintains the Glomerular Filtration Barrier Independent of Mitochondrial Metabolism and Dynamics. Cell Rep. 2019, 27, 1551.e5–1566.e5.
- Zhang, T.; Chi, Y.; Kang, Y.; Lu, H.; Niu, H.; Liu, W.; Li, Y. Resveratrol ameliorates podocyte damage in diabetic mice via SIRT1/PGC-1alpha mediated attenuation of mitochondrial oxidative stress. J. Cell Physiol. 2019, 234, 5033–5043.
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46.
- Tiranti, V.; Savoia, A.; Forti, F.; D’Apolito, M.F.; Centra, M.; Rocchi, M.; Zeviani, M. Identification of the gene encoding the human mitochondrial RNA polymerase (h-mtRPOL) by cyberscreening of the Expressed Sequence Tags database. Hum. Mol. Genet. 1997, 6, 615–625.
- Falkenberg, M.; Gaspari, M.; Rantanen, A.; Trifunovic, A.; Larsson, N.G.; Gustafsson, C.M. Mitochondrial transcription factors B1 and B2 activate transcription of human mtDNA. Nat. Genet. 2002, 31, 289–294.
- Surovtseva, Y.V.; Shutt, T.E.; Cotney, J.; Cimen, H.; Chen, S.Y.; Koc, E.C.; Shadel, G.S. Mitochondrial ribosomal protein L12 selectively associates with human mitochondrial RNA polymerase to activate transcription. Proc. Natl. Acad. Sci. USA 2011, 108, 17921–17926.
- Gu, X.; Liu, Y.; Wang, N.; Zhen, J.; Zhang, B.; Hou, S.; Cui, Z.; Wan, Q.; Feng, H. Transcription of MRPL12 regulated by Nrf2 contributes to the mitochondrial dysfunction in diabetic kidney disease. Free. Radic. Biol. Med. 2021, 164, 329–340.
- Thome, T.; Kumar, R.A.; Burke, S.K.; Khattri, R.B.; Salyers, Z.R.; Kelley, R.C.; Coleman, M.D.; Christou, D.D.; Hepple, R.T.; Scali, S.T.; et al. Impaired muscle mitochondrial energetics is associated with uremic metabolite accumulation in chronic kidney disease. JCI Insight 2020, 6, e139826.
- Szeto, H.H.; Liu, S.; Soong, Y.; Seshan, S.V.; Cohen-Gould, L.; Manichev, V.; Feldman, L.C.; Gustafsson, T. Mitochondria Protection after Acute Ischemia Prevents Prolonged Upregulation of IL-1beta and IL-18 and Arrests CKD. J. Am. Soc. Nephrol. 2017, 28, 1437–1449.
- Lin, H.Y.; Chen, Y.; Chen, Y.H.; Ta, A.P.; Lee, H.C.; MacGregor, G.R.; Vaziri, N.D.; Wang, P.H. Tubular mitochondrial AKT1 is activated during ischemia reperfusion injury and has a critical role in predisposition to chronic kidney disease. Kidney Int. 2021, 99, 870–884.
- Bartoszewska, S.; Collawn, J.F. Unfolded protein response (UPR) integrated signaling networks determine cell fate during hypoxia. Cell Mol. Biol. Lett. 2020, 25, 18.
- Walter, F.; O’Brien, A.; Concannon, C.G.; Dussmann, H.; Prehn, J.H.M. ER stress signaling has an activating transcription factor 6alpha (ATF6)-dependent “off-switch”. J. Biol. Chem. 2018, 293, 18270–18284.
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021, 21, 548–569.
- Huang, J.; Pan, H.; Wang, J.; Wang, T.; Huo, X.; Ma, Y.; Lu, Z.; Sun, B.; Jiang, H. Unfolded protein response in colorectal cancer. Cell Biosci. 2021, 11, 26.

