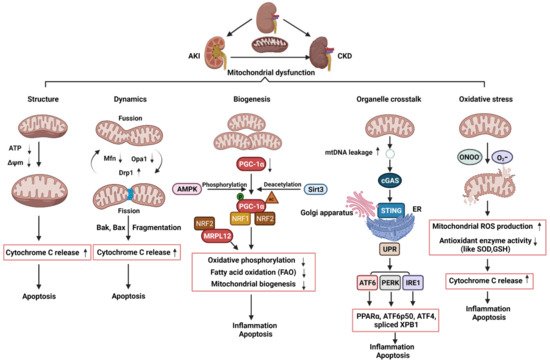
Mitochondria are heterogeneous and highly dynamic organelles, playing critical roles in adenosine triphosphate (ATP) synthesis, metabolic modulation, reactive oxygen species (ROS) generation, and cell differentiation and death. Mitochondrial dysfunction has been recognized as a contributor in many diseases. The kidney is an organ enriched in mitochondria and with high energy demand in the human body. Recent studies have been focusing on how mitochondrial dysfunction contributes to the pathogenesis of different forms of kidney diseases, including acute kidney injury (AKI) and chronic kidney disease (CKD). AKI has been linked to an increased risk of developing CKD. AKI and CKD have a broad clinical syndrome and a substantial impact on morbidity and mortality, encompassing various etiologies and representing important challenges for global public health. Renal mitochondrial disorders are a common feature of diverse forms of AKI and CKD, which result from defects in mitochondrial structure, dynamics, and biogenesis as well as crosstalk of mitochondria with other organelles. Persistent dysregulation of mitochondrial homeostasis in AKI and CKD affects diverse cellular pathways, leading to an increase in renal microvascular loss, oxidative stress, apoptosis, and eventually renal failure. It is important to understand the cellular and molecular events that govern mitochondria functions and pathophysiology in AKI and CKD, which should facilitate the development of novel therapeutic strategies. This review provides an overview of the molecular insights of the mitochondria and the specific pathogenic mechanisms of mitochondrial dysfunction in the progression of AKI, CKD, and AKI to CKD transition. We also discuss the possible beneficial effects of mitochondrial-targeted therapeutic agents for the treatment of mitochondrial dysfunction-mediated AKI and CKD, which may translate into therapeutic options to ameliorate renal injury and delay the progression of these kidney diseases.
Mitochondria are heterogeneous and highly dynamic organelles, playing critical roles in adenosine triphosphate (ATP) synthesis, metabolic modulation, reactive oxygen species (ROS) generation, and cell differentiation and death. Mitochondrial dysfunction has been recognized as a contributor in many diseases. The kidney is an organ enriched in mitochondria and with high energy demand in the human body. Recent studies have been focusing on how mitochondrial dysfunction contributes to the pathogenesis of different forms of kidney diseases, including acute kidney injury (AKI) and chronic kidney disease (CKD). AKI has been linked to an increased risk of developing CKD. AKI and CKD have a broad clinical syndrome and a substantial impact on morbidity and mortality, encompassing various etiologies and representing important challenges for global public health. Renal mitochondrial disorders are a common feature of diverse forms of AKI and CKD, which result from defects in mitochondrial structure, dynamics, and biogenesis as well as crosstalk of mitochondria with other organelles. Persistent dysregulation of mitochondrial homeostasis in AKI and CKD affects diverse cellular pathways, leading to an increase in renal microvascular loss, oxidative stress, apoptosis, and eventually renal failure. It is important to understand the cellular and molecular events that govern mitochondria functions and pathophysiology in AKI and CKD, which should facilitate the development of novel therapeutic strategies. This review provides an overview of the molecular insights of the mitochondria and the specific pathogenic mechanisms of mitochondrial dysfunction in the progression of AKI, CKD, and AKI to CKD transition. We also discuss the possible beneficial effects of mitochondrial-targeted therapeutic agents for the treatment of mitochondrial dysfunction-mediated AKI and CKD, which may translate into therapeutic options to ameliorate renal injury and delay the progression of these kidney diseases.- Chronic kidney disease
- Acute kidney injury
1. The Roles of Mitochondrial in Acute kidney injury, Chronic kidney disease , and Acute kidney injury to Chronic kidney disease Transition
21.1. The Roles of Mitochondrial Structure, Dynamics, and Biogenesis in AKI