Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biology
Mitochondria are heterogeneous and highly dynamic organelles, playing critical roles
in adenosine triphosphate (ATP) synthesis, metabolic modulation, reactive oxygen species (ROS)
generation, and cell differentiation and death. Mitochondrial dysfunction has been recognized as a
contributor in many diseases. The kidney is an organ enriched in mitochondria and with high energy
demand in the human body. Recent studies have been focusing on how mitochondrial dysfunction
contributes to the pathogenesis of different forms of kidney diseases, including acute kidney injury
(AKI) and chronic kidney disease (CKD). AKI has been linked to an increased risk of developing
CKD. AKI and CKD have a broad clinical syndrome and a substantial impact on morbidity and
mortality, encompassing various etiologies and representing important challenges for global public
health. Renal mitochondrial disorders are a common feature of diverse forms of AKI and CKD,
which result from defects in mitochondrial structure, dynamics, and biogenesis as well as crosstalk
of mitochondria with other organelles. Persistent dysregulation of mitochondrial homeostasis in
AKI and CKD affects diverse cellular pathways, leading to an increase in renal microvascular loss,
oxidative stress, apoptosis, and eventually renal failure. It is important to understand the cellular
and molecular events that govern mitochondria functions and pathophysiology in AKI and CKD,
which should facilitate the development of novel therapeutic strategies. This review provides an
overview of the molecular insights of the mitochondria and the specific pathogenic mechanisms of
mitochondrial dysfunction in the progression of AKI, CKD, and AKI to CKD transition. We also
discuss the possible beneficial effects of mitochondrial-targeted therapeutic agents for the treatment
of mitochondrial dysfunction-mediated AKI and CKD, which may translate into therapeutic options
to ameliorate renal injury and delay the progression of these kidney diseases.
- Chronic kidney disease
- Acute kidney injury
1. The Roles of Mitochondrial in Acute kidney injury, Chronic kidney disease , and Acute kidney injury to Chronic kidney disease Transition
Mitochondria are very sensitive to changes in environmental factors, which may cause mitochondrial dysfunction [39]. Mitochondrial dysfunction may result in a decrease of ATP generation, an increase of reactive oxygen species level and an induction of apoptosis, all of which contribute to the development and progression of Acute kidney injury (AKI) and Chronic kidney disease (CKD) as well as AKI to CKD transition. Therefore, it is important to understand the roles of mitochondrial biology and pathophysiology in AKI and CKD, which should facilitate novel discoveries for effective therapies of these diseases.
1.1. The Roles of Mitochondrial Structure, Dynamics, and Biogenesis in AKI
Mitochondria has been recognized as a critical player in AKI with dual roles as the primary source of energy for each cell and as a key regulator of cell death. First, the changes of mitochondria structure have been observed in AKI. Ischemia as a leading cause of AKI diminishes the amounts of mitochondria and induces mitochondria structural changes, typically swelling and showing the disappearance of the inter mitochondrial membrane cristae, due to ATP depletion and membrane potential reduction [40] (Figure 1). In addition, the opening of mitochondrial permeability transition pores (mPTP) due to mitochondrial swelling and dysfunction is a key event that contributes to AKI progression through releasing pro-apoptotic mediators, including cytochrome c, which can induce renal cell apoptosis [41]. mPTP is a nonspecific channel for signal transduction or material transfer between mitochondrial matrix and cytoplasm. In ADR-induced nephropathy rats, treatment with mPTP inhibitor cyclosporine A (CSA) significantly inhibited cytochrome c release and cell apoptosis as well as improved mitochondrial function [42].
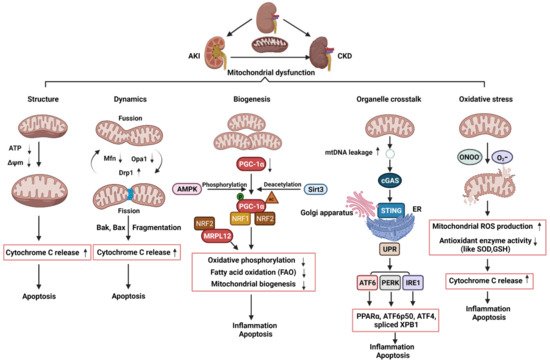
Figure 1. Schematic illustration of pathophysiological processes of mitochondrial dysfunction, including alterations of mitochondrial structure, dynamics, biogenesis, organelle crosstalk, and oxidative stress, in the AKI, CKD, and AKI to CKD transition. AKI: acute kidney injury; CKD: chronic kidney disease; ATP: adenosine triphosphate; ΔΨm: mitochondrial membrane potential; Mfn1: mitofusin 1; Opa1: optic atrophy 1; Drp1: dynamin related protein 1; mtDNA: mitochondria DNA; AMPK: AMP-activated protein kinase; PGC-1α: PPARgamma-coactivator-1α; Sirt3: sirtuin 3; NRF1: nuclear respiratory factor 1; NRF2: nuclear respiratory factor 2; MRPL12: mitochondrial ribosomal protein L12; cGAS; cyclic guanosine monophosphate–adenosine monophosphate (GMP–AMP) synthase; STING: stimulator of interferon genes; ER: endoplasmic reticulum; UPR: unfolded protein response; PPARα: peroxisome proliferator–activated receptor-α; IRE1: inositol-requiring enzyme 1; PERK: PRKR-like ER kinase; ATF6α activating transcription factor 6α; XPB1: the X-box binding protein 1; eIF2α: eukaryotic initiation factor 2α; ATF4: activating transcription factor 4; ONOO-: peroxynitrite; O2-:superoxide; SOD: superoxide dismutase; GSH: glutathione; ROS: reactive oxygen species.
Second, the disruption of the balance between fission and fusion events has been associated with AKI progression. The protein Drp1 that could regulate mitochondrial fission was rapidly activated while the proteins, Mfn and Opa1, which could regulate mitochondrial fusion, were decreased following AKI, resulting in mitochondrial fragmentation [43,44] (Figure 1). Cellular stress leads to the oligomerization of Bax and Bak, two proteins of the pro-apoptotic Bcl-2 family, which are susceptible to insert into fragmented mitochondria and consequently outer membrane permeabilization [45]. The permeabilization of mitochondrial outer membrane (MOMP) by pro-apoptotic Bcl-2 family proteins results in the release of apoptogenic factors, such as cytochrome c, which further bind apoptotic peptidase activating factor 1 (Apaf-1) to recruit and activate caspase 9 to trigger the intrinsic apoptotic pathway [46,47,48] (Figure 1). Knockout of Bax or Bak prevented mitochondrial fragmentation along with suppressed cytochrome c release in AKI [49]. Inhibition of mitochondrial fragmentation pharmacologically or by genetically preventing inflammation and cell death shows a significant renoprotective effect in AKI model induced by ischemic reperfusion or nephrotoxic [44,49,50,51].
Third, the dysregulation of mitochondrial biogenesis has also been observed in AKI. In kidneys, PGC-1α is predominantly expressed in proximal tubules and drives mitochondrial biogenesis by its transcriptional co-activators, such as nuclear respiratory factor 1 (NRF1) and nuclear respiratory factor 2 (NRF2). The target genes of NRF1/2 regulate oxidative phosphorylation, fatty acid oxidation (FAO), and the biogenesis of nicotinamide adenine dinucleotide (NAD+), a central metabolic coenzyme/cosubstrate involved in cellular energy, which refer to oxidative metabolism to renal protection [52,53,54,55] (Figure 1). PGC-1α coordinately upregulates the enzymes that synthesize NAD de novo from amino acids, whereas PGC-1α deficiency or AKI attenuates the de novo pathway. The effect of PGC-1α and its association with mitochondrial biogenesis in AKI was supported by the results generated from in Pgc1α-/- mice following ischemia-reperfusion injury [54]. In cisplatin-induced AKI, treatment with AMPK activator 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR) and the antioxidant agent acetyl-l-carnitine (ALCAR), which activates sirtuin3 (SIRT3) increased PGC-1α activity and improves renal function [56]. Global or tubule-specific PGC-1α knockout mice had normal basal renal function but suffered persistent injury due to prolonged sepsis [57]. In sepsis-associated AKI, endotoxic insults selectively suppress the expression of PGC-1α and then affected PGC-1a mediating the recruitment of NRF1 and NRF2 on genes that regulate oxidative phosphorylation [57,58] (Figure 1). Mitochondrial FAO in proximal tubular cells is a major source of ATP generation. The impairment of mitochondrial FAO has been linked to ATP depletion-induced AKI, and its long-term sequelae leads to CKD [59]. In cisplatin-induced AKI, downregulation of PGC-1α decreased the transcription of FAO genes, including carnitine O-palmitoyltransferase and medium chain–specific acyl-CoA dehydrogenase, leading to decreased mitochondrial FAO (Figure 1). Together, these pieces of evidence indicate that kidney repair or recovery from AKI is associated with mitochondrial structure, dynamics, and biogenesis.
1.2. The Roles of Mitochondrial Dynamics and Biogenesis in CKD
Defective mitochondrial dynamics plays an important role in CKD [60]. In experimental models of diabetic kidney disease (DKD) and in kidney biopsy subjects from patients with DKD, tubular cells and podocytes showed an increase in mitochondrial fragmentation [61,62]. In DKD mice, Drp1 was phosphorylated at serine 600 (p-Drp1S600) and mutation of this serine to alanine exhibited improved biochemical and histological features of diabetic nephropathy. Drp1S600 mutation reduced mitochondrial fission and diminished mitochondrial reactive oxygen species (mtROS), further highlighting the stimulus-specific consequences of Drp1 Serine 600 phosphorylation in mitochondrial fission and progression of DKD [63]. Consistent with these findings, pharmacological inhibition, or knockout of Drp1 ameliorated DKD progression as seen with reduced albuminuria, mesangial matrix expansion, and improved podocyte foot process [64,65]. In unilateral ureter obstruction (UUO) mice, Drp1 was phosphorylated at serine 616 (p-Drp1S616) and pharmacological inhibition of mitochondrial fission reduced fibroblasts accumulation, and interstitial fibrosis along with decreased mitochondrial fragmentation and mitochondrial ROS, suggesting that inhibition of the phospho-Drp1S616-mediated mitochondrial fission attenuated fibroblast activation and proliferation in renal fibrosis [66]. Altogether, these findings suggest that mitochondrial fragmentation, owing to a loss of mitochondrial dynamics, plays a critical role in the development of CKD and may serve as a therapeutic target for retarding CKD progression.
Defective mitochondrial biogenesis also plays an important role in CKD. The expression of PGC-1α was decreased not only in experimental CKD models, but also in kidneys from CKD patients [67,68,69,70,71]. Moreover, PGC-1α and PGC-1α-dependent mitochondrial gene expression positively correlated with the glomerular filtration rate and negatively correlated with fibrosis [71]. In DKD, deletion of PGC-1α in podocytes significantly reduced mtDNA, which demonstrates the impact of PGC-1α on mitochondrial biogenesis in podocytes. [72] (Figure 2). Transgenic expression of PGC-1α or activation of PGC1-α in both experimental diabetic nephropathy and cultured podocytes decreased diabetes-induced podocytopenia and glomerular oxidative stress along with prevented mitochondrial dysfunction and cell death [68,73]. PGC-1α was also downregulated in different murine models of renal fibrosis, including Notch transgenic mice and folic acid treatment mice. Tubule-specific overexpression of PGC-1α in these mice result in reducing fibrosis and restoring mitochondrial content [71,74]. In addition to PGC-1α, other additional factors have also been proposed to contribute to altered mitochondrial bioenergetics in CKD, including deletions and mutations of mtDNA and the changes of lipid composition of mitochondrial membranes [74]. The transcription of human mtDNA in vitro requires the single-subunit mitochondrial RNA polymerase (POLRMT) and human mitochondrial transcription factor B2, h-mtTFB2 [75,76]. Mitochondrial ribosomal protein L12 (MRPL12) binds and activates POLRMT to positively control the mitochondrial oxidative phosphorylation and mtDNA copy number [77]. In diabetic kidneys, the expression of MRPL12 was decreased, which was correlated with alterations of mitochondrial function via NRF2 signaling pathway [78] (Figure 2). In addition, a recent study in CKD indicates that reduced activity of mitochondrial matrix dehydrogenases in the skeletal muscle leads to mitochondrial oxidative phosphorylation dysfunction, which correlates with glomerular filtration rate [79]. Further analysis revealed the accumulation of uremic toxins in the muscle that was strongly associated with the degree of mitochondrial impairment. In sum, altered mitochondrial biogenesis has an important effect on the development and progression of CKD.
1.3. The Roles of Mitochondrial Dysfunction and Its Crosstalk with ER in the Transition of AKI to CKD
Damaged mitochondria release harmful molecules, such as ROS, DNA, and cardiolipin, which can activate NOD-like receptors (NLR) and elevate the levels of proinflammatory cytokines and chemokines, such as IL-18 and IL-1β, to induce persistent renal injury [80] (Figure 1). The innate immune system has been implicated in both AKI and CKD and persistent inflammation after AKI prevents tissue repair and tubular apoptosis. On one hand, inflammation plays a critical role in the initiation and progression of renal fibrosis, when mitochondrial damage persists long after ischemia to sustain chronic inflammasome activation, leading to persistent endothelial injury, podocyte damage, microvascular rarefaction, and ultimately, progressive glomerular and interstitial fibrosis. On the other hand, upon kidney injury, oxidant stress, abundant cytokines, or hypoxia, deteriorate the mitochondrial membrane potential by excreting ROS and releasing pro-apoptotic factors, such as cytochrome c and apoptosis-inducing factor (AIF), which promote caspase dependent and independent apoptosis. In this perspective, persistent mitochondrial dysfunction result in persistent tubular damage, which may affect renal recovery from AKI and further progression to CKD. In a study with AKI to CKD transition experimental model, the investigators performed a long (nine months) follow-up, exploring the role of mitochondria in rats [80]. They confirmed that AKI is not merely an acute phenomenon but results in long-lasting morphologic and functional consequences. AKI induced peritubular and glomerular capillary loss, podocyte damage, and increased profibrotic and proinflammatory cytokines from one to nine months, leading to progressive glomerular and interstitial fibrosis. Transmission electron microscopy revealed major alterations of mitochondria including loss of cristae and matrix density in endothelial cells, podocytes, and tubular cells up to nine months after the injury. Similar data was also presented in the Lan et al. study [40], which showed that persistence in mitochondrial morphologic alterations and significant reductions in mitochondrial number and metabolic dysfunctions at 14 days after IRI plays a key role in the development of renal tubular atrophy and the transition to CKD after AKI. Studies have also focused on the role and mechanisms of impaired protein kinase B (PKB/AKT1) signaling, which works together with mitochondrial proteins, in the regulation of ATP production and oxidative phosphorylation in renal tubular epithelial cells. Mitochondrial AKT1 inhibition led to activation of caspases and tubular cell death, and renal fibrosis after ischemia-reperfusion injury [81]. Altogether, these studies suggest long-term mitochondrial damage may affect the pathophysiology and recovery from AKI and result in the gradual progression to CKD.
In addition, studies show that mitochondrial dysfunction disrupts the crosstalk between mitochondria and ER, leading to tubular inflammation and fibrosis as well as the AKI to CKD transition. ER is a major organelle that controls protein synthesis, folding, and degradation via the unfolded protein response (UPR) pathway. The UPR promotes cellular survival by restoring ER and mitochondrial homeostasis through distinct signaling networks, but if unsuccessful, the UPR induces cell death [82]. UPR pathways is regulated by three distinctive transmembrane sensors: activating transcription factor 6 (ATF6), PRKR-like ER kinase (PERK), and inositol-requiring enzyme 1 (IRE1), which can be activated under ER stress [83] (Figure 1). Various types of kidney damage are associated with dysfunction of the ER and the activation of the UPR. In cisplatin-induced AKI model, cisplatin-induced mitochondrial damage and mtDNA leakage into the cytosol in renal tubular cells, and damaged mtDNA subsequently increased the activation of cyclic guanosine monophosphate–adenosine monophosphate (GMP–AMP) synthase (cGAS)-stimulator of interferon genes (STING) pathway, resulting in the activation of UPR response and then renal inflammation and AKI progression [84]. The activation of ATF6α caused by pathogenic conditions significantly reduces mitochondrial fatty acid β-oxidation activity through suppressing the expression of peroxisome proliferator–activated receptor-α (PPARα) and thereby induced tubular inflammation and fibrosis after acute kidney injury induced by lipotoxicity [70]. Furthermore, activated ATF6 can be translocated to the Golgi apparatus for cleavage to form an active fragment (ATF6 p50). The activation of IRE1 and PERK induces the splicing of the X-box binding protein 1 (XBP1) mRNA and phosphorylates eIF2α, which promotes the translation of activating transcription factor 4 (ATF4) and suppresses the translation of other mRNAs to reduce unfolded proteins, respectively. ATF6 p50, spliced XBP1, and ATF4 could induce the transcription of various UPR target genes that regulate inflammation and apoptosis (Figure 1) [85]. These findings suggest that alterations in ER–mitochondria crosstalk may contribute to the progression of AKI to CKD transition.
This entry is adapted from the peer-reviewed paper 10.3390/ijms222011253
This entry is offline, you can click here to edit this entry!