 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Maha Saber-Ayad | + 3005 word(s) | 3005 | 2021-09-28 04:47:01 | | | |
| 2 | Bruce Ren | -13 word(s) | 2992 | 2021-10-13 03:04:17 | | |
Video Upload Options
The most frequent mutated oncogene family in the history of human cancer is the RAS gene family, including NRAS, HRAS, and, most importantly, KRAS. A hallmark of pancreatic cancer, recalcitrant cancer with a very low survival rate, is the prevalence of oncogenic mutations in the KRAS gene. Due to this fact, studying the function of KRAS and the impact of its mutations on the tumor microenvironment (TME) is a priority for understanding pancreatic cancer progression and designing novel therapeutic strategies for the treatment of the dismal disease. Despite some recent enlightening studies, there is still a wide gap in our knowledge regarding the impact of KRAS mutations on different components of the pancreatic TME. In this review, we will present an updated summary of mutant KRAS role in the initiation, progression, and modulation of the TME of pancreatic ductal adenocarcinoma (PDAC).
1. Introduction
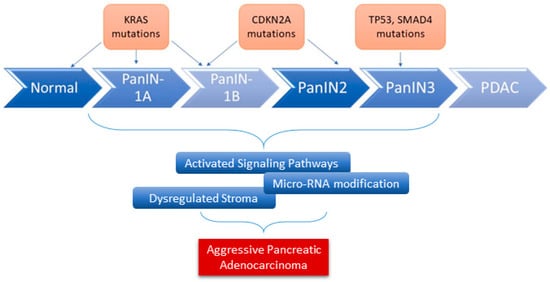
2. Pathobiology of Pancreatic Ductal Adenocarcinoma
3. KRAS Signaling Pathways in PDAC
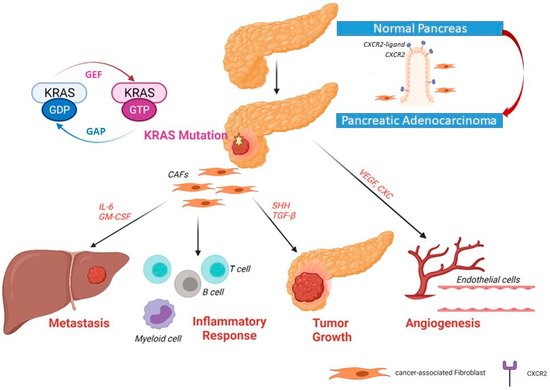
4. Mutated KRAS and the Tumor Microenvironment
4.1. Inflammatory Chemokines, Cytokines, and Interleukin 6
4.2. Mutated KRAS Effect on the Surrounding Stromal Cells
4.3. Mutated KRAS Interaction with the Immune Cells
References
- Bournet, B.; Buscail, C.; Muscari, F.; Cordelier, P.; Buscail, L. Targeting KRAS for diagnosis, prognosis, and treatment of pancreatic cancer: Hopes and realities. Eur. J. Cancer 2016, 54, 75–83.
- di Magliano, M.P.; Logsdon, C.D. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology 2013, 144, 1220–1229.
- Indini, A.; Rijavec, E.; Ghidini, M.; Cortellini, A.; Grossi, F. Targeting KRAS in Solid Tumors: Current Challenges and Future Op-portunities of Novel KRAS Inhibitors. Pharmaceutics 2021, 13, 653.
- Waters, A.M.; Ozkan-Dagliyan, I.; Vaseva, A.V.; Fer, N.; Strathern, L.A.; Hobbs, G.A.; Tessier-Cloutier, B.; Gillette, W.K.; Bagni, R.; Whiteley, G.R.; et al. Evaluation of the selectivity and sensitivity of isoform- and mutation-specific RAS antibodies. Sci. Signal. 2017, 10, eaao3332.
- Maitra, A.; Hruban, R.H. Pancreatic cancer. Annu. Rev. Pathology. 2008, 3, 157–188.
- Sun, H.; Zhang, B.; Li, H. The Roles of Frequently Mutated Genes of Pancreatic Cancer in Regulation of Tumor Microenvi-ronment. Technol. Cancer Res. Treat. 2020, 19, 1533033820920969.
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The Pancreas Cancer Microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276.
- Cancer Genome Atlas Research Network. Electronic address aadhe, Cancer Genome Atlas Research, N. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2017, 32, 185–203.
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.-C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.-M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405.
- Masetti, M.; Acquaviva, G.; Visani, M.; Tallini, G.; Fornelli, A.; Ragazzi, M.; Vasuri, F.; Grifoni, D.; Di Giacomo, S.; Fiorino, S.; et al. Long-term survivors of pancreatic adenocarcinoma show low rates of genetic alterations in KRAS, TP53 and SMAD4. Cancer Biomark. 2018, 21, 323–334.
- Visani, M.; Acquaviva, G.; De Leo, A.; Sanza, V.; Merlo, L.; Maloberti, T.; Brandes, A.A.; Franceschi, E.; Di Battista, M.; Masetti, M.; et al. Molecular alterations in pancreatic tumors. World J. Gastroenterol. 2021, 27, 2710–2726.
- Tanizaki, J.; Okamoto, I.; Sakai, K.; Nakagawa, K. Differential roles of trans-phosphorylated EGFR, HER2, HER3, and RET as heterodimerisation partners of MET in lung cancer with MET amplification. Br. J. Cancer 2011, 105, 807–813.
- Jahng, A.W.; Reicher, S.; Chung, D.; Varela, D.; Chhablani, R.; Dev, A.; Pham, B.; Nieto, J.; Venegas, R.J.; French, S.W.; et al. Staining for p53 and Ki-67 increases the sensitivity of EUS-FNA to detect pancreatic malignancy. World J. Gastrointest. Endosc. 2010, 2, 362–368.
- Hsieh, Y.-Y.; Liu, T.-P.; Chou, C.-J.; Chen, H.-Y.; Lee, K.-H.; Yang, P.-M. Integration of Bioinformatics Resources Reveals the Therapeutic Benefits of Gemcitabine and Cell Cycle Intervention in SMAD4-Deleted Pancreatic Ductal Adenocarcinoma. Genes 2019, 10, 766.
- Bartsch, D.; Shevlin, D.W.; Tung, W.S.; Kisker, O.; Wells, S.A., Jr.; Goodfellow, P.J. Frequent mutations ofCDKN2 in primary pancreatic adenocarcinomas. Genes Chromosom. Cancer 1995, 14, 189–195.
- Rozenblum, E.; Schutte, M.; Goggins, M.; Hahn, S.; Panzer, S.; Zahurak, M.; Goodman, S.N.; Sohn, T.A.; Hruban, R.H.; Yeo, C.J.; et al. Tumor-suppressive pathways in pancreatic carcinoma. Cancer Res. 1997, 57, 1731–1734.
- Oshima, M.; Okano, K.; Muraki, S.; Haba, R.; Maeba, T.; Suzuki, Y.; Yachida, S. Immunohistochemically Detected Expression of 3 Major Genes (CDKN2A/p16, TP53, and SMAD4/DPC4) Strongly Predicts Survival in Patients with Resectable Pancreatic Cancer. Ann. Surg. 2013, 258, 336–346.
- Qian, Y.; Gong, Y.; Fan, Z.; Luo, G.; Huang, Q.; Deng, S.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. Molecular alterations and targeted therapy in pancreatic ductal ade-nocarcinoma. J. Hematol. Oncol. 2020, 13, 130.
- Wong, W.; Raufi, A.G.; Safyan, R.A.; Bates, S.E.; Manji, G.A. BRCA Mutations in Pancreas Cancer: Spectrum, Current Management, Challenges and Future Prospects. Cancer Manag. Res. 2020, 12, 2731–2742.
- Khan, M.A.; Azim, S.; Zubair, H.; Bhardwaj, A.; Patel, G.K.; Khushman, M.; Singh, S.; Singh, A.P. Molecular Drivers of Pancreatic Cancer Patho-genesis: Looking Inward to Move Forward. Int. J. Mol. Sci. 2017, 18, 779.
- Kleeff, J.; Korc, M.; Apte, M.; Vecchia, C.L.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022.
- Consortium TAPG. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831.
- Lu, S.; Jang, H.; Nussinov, R.; Zhang, J. The Structural Basis of Oncogenic Mutations G12, G13 and Q61 in Small GTPase K-Ras4B. Sci. Rep. 2016, 6, 21949.
- Cicenas, J.; Kvederaviciute, K.; Meskinyte, I.; Meskinyte-Kausiliene, E.; Skeberdyte, A. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 Mutations in Pancreatic Cancer. Cancers 2017, 9, 42.
- Zeitouni, D.; Pylayeva-Gupta, Y.; Der, C.J.; Bryant, K.L. KRAS Mutant Pancreatic Cancer: No Lone Path to an Effective Treatment. Cancers 2016, 8, 45.
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774.
- Jonckheere, N.; Vasseur, R.; Van Seuningen, I. The cornerstone K-RAS mutation in pancreatic adenocarcinoma: From cell sig-naling network, target genes, biological processes to therapeutic targeting. Crit. Rev. Oncol. Hematology 2017, 111, 7–19.
- Dey, P.; Li, J.; Zhang, J.; Chaurasiya, S.; Strom, A.; Wang, H.; Liao, W.-T.; Cavallaro, F.; Denz, P.; Bernard, V.; et al. Oncogenic KRAS-Driven Metabolic Reprogramming in Pancreatic Cancer Cells Utilizes Cytokines from the Tumor Microenvironment. Cancer Discov. 2020, 10, 608–625.
- Buscail, L.; Bournet, B.; Cordelier, P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 153–168.
- Liu, P.; Wang, Y.; Li, X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm. Sin. B 2019, 9, 871–879.
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a03145.
- Preis, M.; Korc, M. Signaling pathways in pancreatic cancer. Crit. Rev. Eukaryot. Gene Expr. 2011, 21, 115–129.
- Zhao, S.; Wang, Y.; Cao, L.; Ouellette, M.M.; Freeman, J.W. Expression of oncogenic K-ras and loss of Smad4 cooperate to induce the expression of EGFR and to promote invasion of immortalized human pancreas ductal cells. Int. J. Cancer 2010, 127, 2076–2087.
- Yu, C.; Merza, M.; Luo, L.; Thorlacius, H. Inhibition of Ras signalling reduces neutrophil infiltration and tissue damage in severe acute pancreatitis. Eur. J. Pharmacol. 2015, 746, 245–251.
- Matsuo, Y.; Campbell, P.M.; Brekken, R.A.; Sung, B.; Ouellette, M.M.; Fleming, J.B.; Aggarwal, B.B.; Der, C.; Guha, S. K-Ras Promotes Angiogenesis Mediated by Immortalized Human Pancreatic Epithelial Cells through Mitogen-Activated Protein Kinase Signaling Pathways. Mol. Cancer Res. 2009, 7, 799–808.
- Kitajima, S.; Thummalapalli, R.; Barbie, D.A. Inflammation as a driver and vulnerability of KRAS mediated oncogenesis. Semin. Cell Dev. Biol. 2016, 58, 127–135.
- Awaji, M.; Saxena, S.; Wu, L.; Prajapati, D.R.; Purohit, A.; Varney, M.L.; Kumar, S.; Rachagani, S.; Ly, Q.P.; Jain, M.; et al. CXCR2 signaling promotes secretory cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. FASEB J. 2020, 34, 9405–9418.
- Kumar, V.; Donthireddy, L.; Marvel, D.; Condamine, T.; Wang, F.; Lavilla-Alonso, S.; Hashimoto, A.; Vonteddu, P.; Behera, R.; Goins, M.A.; et al. Cancer-Associated Fibroblasts Neutralize the Anti-tumor Effect of CSF1 Receptor Blockade by Inducing PMN-MDSC Infiltration of Tumors. Cancer Cell 2017, 32, 654–668.
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598.
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 2019, 10, 1835.
- Nielsen, M.F.B.; Mortensen, M.B.; Detlefsen, S. Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells. World J. Gastroenterol. 2016, 22, 2678–2700.
- Mei, L.; Du, W.; Ma, W.W. Targeting stromal microenvironment in pancreatic ductal adenocarcinoma: Controversies and promises. J. Gastrointest. Oncol. 2016, 7, 487–494.
- Hamarsheh, S.; Groß, O.; Brummer, T.; Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 2020, 11, 5439.
- Wang, T.; Notta, F.; Navab, R.; Joseph, J.; Ibrahimov, E.; Xu, J.; Zhu, C.-Q.; Borgida, A.; Gallinger, S.; Tsao, M.-S. Senescent Carcinoma-associated Fibroblasts Upregulate IL8 to Enhance Pro-metastatic Phenotypes. Mol. Cancer Res. 2016, 15, 3–14.
- Aldinucci, D.; Borghese, C.; Casagrande, N. The CCL5/CCR5 Axis in Cancer Progression. Cancers 2020, 12, 1765.
- Singh, S.K.; Mishra, M.K.; Eltoum, I.-E.A.; Bae, S.; Lillard, J.W., Jr.; Singh, R. CCR5/CCL5 axis interaction promotes migratory and invasiveness of pancreatic cancer cells. Sci. Rep. 2018, 8, 1323.
- Huffman, A.P.; Lin, J.H.; Kim, S.I.; Byrne, K.T.; Vonderheide, R.H. CCL5 mediates CD40-driven CD4+ T cell tumor infiltration and immunity. JCI Insight 2020, 5, e137263.
- Veatch, J.R.; Jesernig, B.L.; Kargl, J.; Fitzgibbon, M.; Lee, S.M.; Baik, C.; Martins, R.; Hughton, M.; Riddell, S.R. Endogenous CD4(+) T Cells Recognize Neoantigens in Lung Cancer Patients, Including Recurrent Oncogenic KRAS and ERBB2 (Her2) Driver Mutations. Cancer Immunol. Res. 2019, 7, 910–922.
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 2140–2141.
- Lesina, M.; Kurkowski, M.U.; Ludes, K.; Rose-John, S.; Treiber, M.; Klöppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akira, S.; et al. Stat3/Socs3 Activation by IL-6 Transsignaling Promotes Progression of Pancreatic Intraepithelial Neoplasia and Development of Pancreatic Cancer. Cancer Cell 2011, 19, 456–469.
- Zhang, Y.; Yan, W.; Collins, M.A.; Bednar, F.; Rakshit, S.; Zetter, B.R.; Stanger, B.Z.; Chung, I.; Rhim, A.D.; Di Magliano, M.P. Interleukin-6 Is Required for Pancreatic Cancer Progression by Promoting MAPK Signaling Activation and Oxidative Stress Resistance. Cancer Res. 2013, 73, 6359–6374.
- Tape, C.J.; Ling, S.; Dimitriadi, M.; McMahon, K.M.; Worboys, J.D.; Leong, H.S.; Norre, I.C.; Miller, C.J.; Poulogiannis, G.; Laffenburger, D.A.; et al. Oncogenic KRAS Regulates Tumor Cell Signaling via Stromal Reciprocation. Cell 2016, 165, 1818.
- Gu, D.; Schlotman, K.E.; Xie, J. Deciphering the role of hedgehog signaling in pancreatic cancer. J. Biomed. Res. 2016, 30, 353–360.
- Phillips, P. Pancreatic stellate cells and fibrosis. In Pancreatic Cancer and Tumor Microenvironment; Grippo, P.J., Munshi, H.G., Eds.; Transworld Research Network: Trivandrum, India, 2012; Chapter 3.
- Jonckheere, N.; Van Seuningen, I. The membrane-bound mucins: From cell signalling to transcriptional regulation and expression in epithelial cancers. Biochimie 2010, 92, 1–11.
- Vasseur, R.; Skrypek, N.; Duchêne, B.; Renaud, F.; Martínez-Maqueda, D.; Vincent, A.; Porchet, N.; Van Seuningen, I.; Jonckheere, N. The mucin MUC4 is a transcriptional and post-transcriptional target of K-ras oncogene in pancreatic cancer. Implication of MAPK/AP-1, NF-κB and RalB signaling pathways. Biochim. Biophys. Acta Bioenerg. 2015, 1849, 1375–1384.
- Pommier, A.; Anaparthy, N.; Memos, N.; Kelley, Z.L.; Gouronnec, A.; Yan, R.; Auffray, C.; Albrengues, J.; Egeblad, M.; Iacobuzio-Donahue, C.A.; et al. Unresolved endoplasmic reticulum stress engenders immune-resistant, latent pancreatic cancer metastases. Science 2018, 360, eaao4908.
- Yamamoto, K.; Venida, A.; Yano, J.; Biancur, D.E.; Kakiuchi, M.; Gupta, S.; Sohn, A.S.W.; Mukhopadhyay, S.; Lin, E.Y.; Parker, S.J.; et al. Autophagy Promotes Immune Evasion of Pancreatic Cancer by Degrading MHC-I; Springer: Berlin/Heidelberg, Germany, 2020; Volume 581, ISBN 0000007587.
- Kaur, K.; Chang, H.-H.; Topchyan, P.; Cook, J.M.; Barkhordarian, A.; Eibl, G.; Jewett, A. Deficiencies in Natural Killer Cell Numbers, Expansion, and Function at the Pre-Neoplastic Stage of Pancreatic Cancer by KRAS Mutation in the Pancreas of Obese Mice. Front. Immunol. 2018, 9, 1229.

