1. Introduction
Mutations in the RAS gene family are common in many cancer types. The point mutations in the Kirsten rat sarcoma viral oncogene homolog (
KRAS) gene typically affect the hotspots at codons 12 and 13 [
1,
2] However, at lower frequencies, KRAS mutations can also occur in codons 18, 61, 117, and 146. RAS GTPase-activating protein 1 (is a small guanosine triphosphatase (GTPase) that acts as a molecular switch and interacts with more than 20 effector proteins through localization to the inner surface of the cell membrane [
1,
2]. The point mutation in
KRAS can impair the intrinsic GTPase activity of KRAS protein, preventing its conversion from an active form “guanosine triphosphate” (GTP) to its inactive form “guanosine diphosphate” (GDP). Consequently, KRAS remains permanently bound to GTP resulting in activation of downstream signaling pathways [
1,
2].
KRAS mutations are predominant in most cancers, such as pancreatic ductal adenocarcinoma (PDAC) (86%), colorectal cancer (CRC) (85%), and lung cancer (30%) [
3]. This is followed by
NRAS (12%) mutations, which are predominant in cutaneous melanoma and acute myelogenous leukemia. However,
HRAS mutations that are found in bladder and head and neck squamous cell carcinomas are infrequently seen in other types of cancers [
4]. According to the COSMIC v94 database, 99% of KRAS mutations are missense mutations, mainly with a gain of function.
In this review, we will first discuss the pathobiology of PDAC. Then, the significance of KRAS mutations in PDAC will be discussed. In addition, we will show how modulation of the immune response and promotion of angiogenesis by oncogenic KRAS can alter the tumor microenvironment (TME). We will finally highlight the link between diabetes and PDAC, as well as the importance of vitamin D for effective targeted therapies.
2. Pathobiology of Pancreatic Ductal Adenocarcinoma
Tumors of the exocrine pancreas are, by far, the most common type of pancreatic cancers, of which PDAC is the most common type (90%). PDAC is an epithelial tumor, and its formation requires a stepwise progression over many years. In other words, it requires the transition of a normal pancreatic duct to a pre-invasive precursor lesion, a frank malignant, invasive cancer, then a metastatic tumor. Histologically, there are three morphological noninvasive precursor lesions of PDAC, including pancreatic intraepithelial neoplasms (PanIN), intraductal papillary mucinous neoplasms (IPMN), and mucinous cystic neoplasms (MCN), of which PanIN is the most studied one. PanIN may advance cancer that exhibit invasion, metastasis, and therapeutic resistance through a dense stromal microenvironment (desmoplastic) establishment in addition to the development of genetic variability [
5]. The PDAC TME comprises a myriad of cells in addition to hyaluronic acid, cytokines, chemokines, and a variety of collagens. The cellular component includes macrophages, dendritic cells, T cells, and B cells [
6]. Local immunity is always suppressed, resulting in an ideal milieu for tumor initiation, progression, as well as distant metastasis. The cold tumor with dominant CD4+ regulatory T-cells usually evades the immune system and dense desmoplastic TME hinders the access of therapeutic agents [
7].
Several gene alterations have been identified during tumor progression and interaction with the TME. The whole-exome sequencing analysis of PDAC revealed around 60 genetic alterations, most of which are point mutations [
8]. According to several studies,
KRAS is the most frequently mutated oncogene in PDAC (from 70% to 95%). In addition to
KRAS, mutations were identified in other well-known genes, e.g.,
CDKN2A (encoding p16),
TP53,
ARID1A,
SMAD4, as well as in novel genes, e.g.,
ATM (one of the key genes of DNA repair),
EPC1 and
ARID2 (involved in chromatin modification), and
KDM6A and
PREX2 (involved in carcinogenesis) [
9].
KRAS mutations in exon 3 have a remarkably favorable prognosis. Coexistent
KRAS mutations were detected in the same pancreatic neoplastic mass more frequently than in other tumors.
KRAS mutations coexistent with
TP53 alterations and/or loss of SMAD4 protein herald a worse PDAC prognosis [
10]. The sensitivity of endoscopic ultrasound-guided fine needle aspiration (EUS-FNA) in the diagnosis of pancreatic malignant lesions can be improved by implementing the evaluation of the
TP53 gene [
11].
TP53 alterations have been detected in 50–75% of PDACs. The disease outcome is worsened with loss of normal p53 protein, mainly if combined with
KRAS mutations and loss of expression of SMAD4. The coexisting mutations lead to one of the aberrant signaling nodes in PDAD that shows an enhanced activity of hepatocyte growth factor receptor (HGFR) and its respective tyrosine kinase, epidermal growth factor receptor (EGFR), and an increased expression of neuropilin 1, CD44, and β1 integrin. Such activity is augmented by heterodimerization of HGFR and EGFR [
12]. Approximately 50% of pancreatic cancers harbor inactivated
SMAD4 due to intragenic mutations or homozygous deletion, which occur late in PDAC. The loss of SMAD4 protein is associated with an increased risk of metastases and a worse prognosis [
10,
13]. In PDAC,
SMAD4 mutations result in suppression of TGF-β signal transduction and may lead to altered sensitivity to gemcitabine [
11,
14]. Similarly, approximately 95% of sporadic pancreatic carcinomas have inactivated
CDKN2A as a result of intragenic mutation [
15,
16].
CDKN2A is linked to familial pancreatic cancer. Suppressed p16 expression is associated with larger tumors and with a poorer prognosis [
11,
17]. It is noteworthy that CDK4 inhibitors have shown promising results for the treatment of
CDKN2A-deficient tumors in preclinical PDAC models [
18].
BRCA1/2 mutations have been identified in, 5 to 10% of PDAC. Such mutations may lead to either sporadic or familial disease [
8,
19].
Infrequent genetic alterations and events in PDAC include microsatellite instability (MSI), also known as defective DNA mismatch repair (dMMR), BRAFV600E mutations, and MGMT promoter hypermethylation [
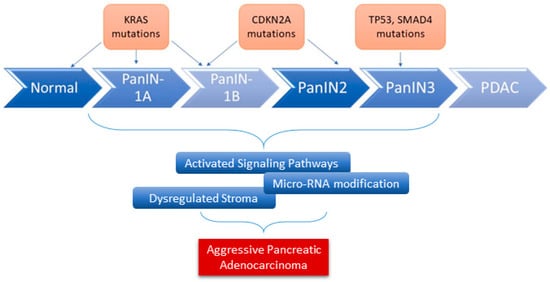
11]. In addition to these genetic alterations, other factors serve as fuel for aggressive pancreatic cancer development. This includes dysregulated stromal-associated factors, signaling pathways, and microRNAs (
Figure 1), [
20].
Figure 1. The role of genes and microRNAs in the progression of PDAC. Alterations in various key genes contribute to the progression of PDAC (overexpression, loss of function mutation, and inactivation). Many other factors have been shown to serve as fuel for the development of aggressive PDAC, including microRNAs dysregulation.
Subgroups of PDAC were defined according to the presence of mutations/genomic alterations/events. Intriguingly, the locally rearranged subgroup is characterized by >50 events limited to one to three chromosomes. These events are typically oncogene amplifications that target existing therapeutics or genomic catastrophes such as chromothripsis [
21].
3. KRAS Signaling Pathways in PDAC
Approximately 86% of somatic alterations in PDAC target
KRAS. G12D and G12V variants account for approximately 80% of
KRAS mutations and hence the initiation of most PDAC cases [
22]. G12 mutation is followed by that of G13 (9%) and Q61 (1%) in PDAC [
23]. Mutations of the
KRAS exon 2 codons G12 and G13 exist in almost all PDAC cases (in more than 95% of PDAC cases). Other mutations, such as Q61 (<1%) in
KRAS exon 3 and K117 and A146 (<1%) in exon 4, seem to be additional hotspots associated with constitutively activated
KRAS in pancreatic cancer [
24].
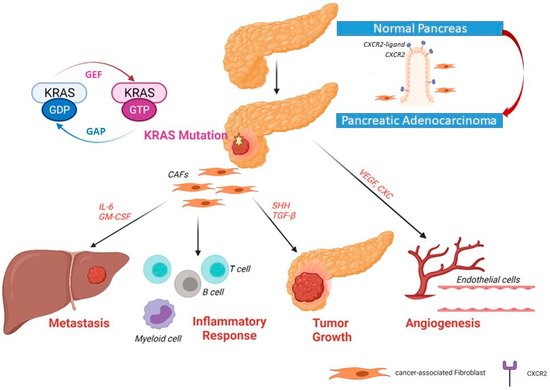
In normal cells, the active state KRAS is bound to GTP, while it is bound to GDP in the inactive state. RAS proteins keep switching “on” and “off” through conformational changes through binding of GTP and GDP. GEF (guanine nucleotide exchange factor) promotes dissociation of GDP and acts as a positive regulator; GAP (GTPase-activating protein) promotes hydrolysis of GTP and acts as a negative regulator helping to keep most of KRAS in an inactive GDP-bound state (
Figure 2) [
25]. Most RAS mutations involve GAP-mediated inactivation of RAS. For example, substitutions in residues G12 prevent van der Waals bond formation between RAS and the GAP, leading to perturbation of Q61 (or the catalytic glutamine) orientation in RAS. This results in the pronounced attenuation of GTP hydrolysis, with enduring activation of RAS-driven downstream pathways [
26]. Activated
KRAS induces a myriad of downstream signaling pathways and effector proteins, such as mitogen-activated protein kinase (MAPK)–MAPK kinase (MEK), phosphoinositide 3-kinase (PI3K)–AKT–the mechanistic target of rapamycin (mTOR), rapidly accelerated fibrosarcoma (RAF)–MEK–extracellular signal-regulated kinase (ERK), and Nuclear factor-κB (NF-κB) pathway (among other nuclear transcription factors). These factors can enhance the survival, proliferation, transformation, and invasion of cancer cells [
27]. Additionally, mutant KRAS results in the autonomous release of type I cytokine complexes by cancer cells. Subsequently, a cascade of events follows that leads to metabolic reprogramming (see
Section 5) [
28]. The signaling pathways of KRAS are discussed comprehensively in previous review articles [
27,
29,
30,
31]. The aforementioned studies point to the potential role of
KRAS mutations in modulating the immune status of the TME.
Figure 2. Upregulated expression of CXCR2 and its ligand in pancreatic ductal adenocarcinoma. In primary pancreatic tumors, the upregulation of CXCR2 expression induces the secretory function in cancer-associated fibroblasts (CAFs) Recruitment of fibroblasts to become CAFs can help tumor cells to grow, induce angiogenesis and invade the portal vein and metastasize to the liver.
4. Mutated KRAS and the Tumor Microenvironment
The modulation of the immune response through several cytokines, as well as the promotion of angiogenesis by oncogenic
KRAS, can alter the TME [
27].
KRAS mutations are likely to coexist with mutations of other genes in PDAC, as previously described. The summative effect on the TME shapes the immune status of the tumor surrounding, a crucial factor that determines the capacity of the tumor to metastasize and to respond to therapeutic agents [
21]. As an example, a worse PDAC prognosis is expected when
KRAS mutations coexist with
TP53 alterations and/or loss of SMAD4 protein. In addition, the combination of
KRAS mutations and loss of SMAD4 enhances the activity of HGFR and EGFR, together with an increased neuropilin 1, CD44, and β1 integrin expression [
21,
32,
33].
Yu and coworkers (2015) showed that RAS signaling regulates pathological inflammation in severe acute pancreatitis. Their study indicated that RAS signaling controls CXC chemokine formation, indirectly affecting neutrophil recruitment and tissue injury in the inflamed pancreatic tissue. Inhibition of RAS signaling resulted in the decreased taurocholate-induced pancreatic activity of myeloperoxidase, which indicates the suppression of neutrophil recruitment [
34]. KRAS was involved in CXC chemokine formation and the induction of VEGF, which plays a critical role in pancreatic angiogenesis. Furthermore, RAS was shown to upregulate COX2, which, in turn, promotes tumor formation via MEK/c-Jun pathway and human umbilical vein endothelial cells (HUVEC) invasion [
34,
35].
4.1. Inflammatory Chemokines, Cytokines, and Interleukin 6
Several chemokines have been implicated in inflammation-induced tumorigenesis. The induction of several inflammatory cytokines and chemokines responsible for tumorigenesis and invasiveness has been tightly linked to oncogenic
KRAS [
36]. C-X-C motif chemokine receptor 2 (CXCR2) controls a major inflammatory signaling network in pancreatic cancers with
KRAS mutation [
37]. Previously, it has been reported that CXCR2, which is a receptor for a group of C-X-C cytokines, can enhance granulocyte recruitment to the site of inflammation, as well as enabling angiogenesis through recruitment of myeloid-derived suppressor cells (MDSCs) and immunosuppressive neutrophils [
38].
KRAS mutations influence the stellate cells/pluripotent stem cells of the pancreas (activated stellate cells are referred to as cancer-associated fibroblasts or CAF). CAFs are one of the key players that promote tumor proliferation, migration, invasion, and metastasis. Furthermore, CAFs modulate the tumor immune microenvironment and modify its response to treatment. Thus, CAFs delineate the acquisition and maintenance of numerous cancer hallmarks [
39,
40]. Recently, it was demonstrated that CXCR2 can induce secretory function in CAFs via NF-κB activation. CAFs make up a united heterogeneous population of cells that can alter the microenvironment of the tumor and thus alter the neoplastic cell’s fate [
37]. CAFs play a major role in the formation of the desmoplastic stroma as well [
41].
CAFs can secrete many extracellular matrix proteins, such as collagen, fibronectin, and laminins, into the TME following their activation [
42]. During carcinogenesis, CAFs can produce inflammatory mediators such as CXCL8 and interleukin-6 (IL-6), both of which are associated with inflammation, tumor growth, and angiogenesis [
43,
44]. Thus, KRAS/CXCR2 signaling plays a major role in regulating the CAFs of PDAC. Another chemokine called chemokine C-C motif ligand 5 (CCL5) is expressed by many cell types such as immune cells, epithelial cells, fibroblasts, and more importantly, by tumor cells [
45]. A study by Singh et al. (2018), showed that the capacity of pancreatic cancer cells to migrate and invade other organs increases via activation of CCR5 by CCL5 that triggers a cascade of signaling pathways [
46]. In another recent study, the authors discovered that CCL5 can mediate the influx of CD4+ T cells into the TME following treatment with CD40 antibody [
47]. CD4+ T cells were shown to have a negative role in tumor immunity and response to immunotherapy [
48]. These studies suggest that therapeutic targeting of inflammatory chemokines might result in improved outcomes in
KRAS mutant cancers.
Interestingly, two key drivers of PDAC tumors, oncogenic
KRAS and hypoxia, have been shown to induce IL-6 [
49]. IL6 secretion has been identified to be the most characterized cytokine in PDAC, which is strongly associated with tumor survival. Its secretion can be induced both by myeloid cells from the surrounding stroma and tumor cells [
50,
51]. Moreover, strong phosphorylation of signal transducer and activator of transcription 3 (STAT3) induced by IL-6 resulted in PanIN-PDAC progression in K-RasG12D mice [
50]. IL-6 has been shown to have a role in tumor formation. According to Zhang et al. (2013), genetic deletion of IL-6 resulted in a reduction in PanIN formation, when
K-Ras mutation was initiated embryonically in an inducible
K-Ras-driven mouse model. The study also showed a significant decrease in the percentage of intra-tumoral cancer-promoting macrophages and MDSCs following the deletion of IL-6 in this
K-Ras-driven PDAC mouse model [
51].
KRAS mutations as therapeutic targets in CAFs will be discussed later in this review [
52].
4.2. Mutated KRAS Effect on the Surrounding Stromal Cells
Tape and coworkers (2016) showed that
KRASG12D communicates with stromal cells and renders tumor cells insensitivity to many important factors. These authors demonstrated that the secretion of growth factor sonic hedgehog (SHH), granulocyte colony-stimulating factor (GCSF), and granulocyte-macrophage colony-stimulating factor (GM-CSF) cytokines can be increased by active oncogenic
KRASG12D [
52]. Hedgehog (Hh) signaling which is known to play a crucial role in embryonic development, stem cell regulation, and adult tissue homeostasis, is highly activated in PDAC [
53]. SHH is a ligand of the hedgehog signaling pathway. An increase in SHH secretion via the NF-κB pathway and
KRAS leads to the disruption of primary cilium of PDAC cells and upregulation of many extracellular matrix components, such as collagen, MMPs, and fibrillin-1. Pancreatic stellate cells (PSCs) cross talk with tumor cells to enhance local tumor growth and promote distant metastasis. It is noteworthy that PSCs represent a major origin of fibrosis in the TME [
54]. SHH can alter the PSC intercellular signaling potential through upregulation of two specific growth factors: insulin-like growth factor 1 (IGF1) and growth arrest-specific gene 6 (GAS6). Via SHH,
KRASG12D PDAC cells can send signals to PSC and, at the same time, remain insensitive to autocrine SHH. This results in further production of IGF1 and GAS6. Consequently, these two growth factors are capable of activating the receptor tyrosine kinases (RTKs), IGF1R, and AXL [
52]. This will eventually lead to increased proliferation, and resistance to apoptosis.
The overexpression of a high molecular weight glycoprotein called Mucin was shown to be associated with progression in many tumors, including PDAC [
55]. Mucin can be divided into two major groups: (1) a membrane-bound mucin called MUC4 that is implicated in cell–cell and cell–extracellular matrix interactions and (2) secreted mucins that participate in epithelial protection [
55]. Interestingly, aberrant activated
KRAS in PDAC can activate and cause upregulation of this membrane-bound mucin MUC4 both at the transcriptional and post-transcriptional level via p42/44 MAPK and NF-κB pathways and RalB pathway, respectively. It has been reported that there is a direct interaction between the promoter of MUC4 with c-Fos (activated by p42/44 MAPK pathway), c-Jun, and p65 NF-κB subunit, suggesting a link between the gradual increase in both
KRAS signaling (MAPK and NF-κB) and MUC4 expression in pancreatic carcinogenesis [
56]. Moreover, silencing of RalB GTPase in PanIN lesions leads to the inhibition of MUC4 protein overexpression with no effect on its mRNA level, whereas RalA silencing has no effect on its protein expression [
56].
4.3. Mutated KRAS Interaction with the Immune Cells
As previously mentioned, PDAC cells harboring mutant
KRAS can secrete chemokines (e.g., GM-CSF and IL-6). These chemokines stimulate various immune cells, including T-cells and B-cells, MDSCs, and macrophages, resulting in an inflammatory TME. In addition, oncogenic KRAS stimulates the release of angiogenic factors (e.g., VEGF) [
27,
29]. These factors can determine the TME immune status, the possibility of tumor metastasis, and the response to treatment.
Immune evasion is a major obstacle to cancer treatment. It was found that PDAC cells lack the expression of cytokeratin 19 (CK19) and display a reduced expression of MHC-I at the cell surface. Additionally, autophagy-related genes were found to be enriched in MHC-I negative PDAC cells that reside in liver metastasis [
57]. In PDAC, surface MHC-I is decreased via the NBR1-mediated autophagy–lysosomal pathway. Recently, it was shown that the surface levels of MHC-I can be restored through inhibition of autophagy [
58]. This inhibition in syngeneic host mice also leads to the enhancement of antitumor T cell responses and consequently reduction in tumor growth.
It has been reported that adipose tissues, in which tumors have a predilection to grow, can convert tumor-suppressive NK cells to tumor-promoting cells through decreasing NK-mediated cytotoxicity and IFN-γ secretion and increasing IL-6 secretion, aiding tumor growth and expansion. According to Kaur et al. (2018), NK cells and monocytes are recruited to the peri-pancreatic and pancreatic adipose tissue from the circulation, where they lose the secretion of IFN-γ, while increasing the secretion of IL-6, thus perpetuating the tumor inflammatory milieu [
59].