Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Adewale Fadaka | + 1546 word(s) | 1546 | 2021-09-10 05:35:51 | | | |
| 2 | Rita Xu | Meta information modification | 1546 | 2021-09-10 10:30:08 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Fadaka, A. Natural Sources against Coronavirus Protease. Encyclopedia. Available online: https://encyclopedia.pub/entry/14070 (accessed on 24 July 2026).
Fadaka A. Natural Sources against Coronavirus Protease. Encyclopedia. Available at: https://encyclopedia.pub/entry/14070. Accessed July 24, 2026.
Fadaka, Adewale. "Natural Sources against Coronavirus Protease" Encyclopedia, https://encyclopedia.pub/entry/14070 (accessed July 24, 2026).
Fadaka, A. (2021, September 10). Natural Sources against Coronavirus Protease. In Encyclopedia. https://encyclopedia.pub/entry/14070
Fadaka, Adewale. "Natural Sources against Coronavirus Protease." Encyclopedia. Web. 10 September, 2021.
Copy Citation
The SARS-CoV-2 main protease (Mpro) is one of the molecular targets for drug design. Effective vaccines have been identified as a long-term solution but the rate at which they are being administered is slow in several countries, and mutations of SARS-CoV-2 could render them less effective.
flavonols
SARS-CoV-2
Mpro
natural products
MDs
MM/GBSA
docking
COVID-19
in silico approach
1. Introduction
Viral infection is one of the major challenges faced by human health, and many viral diseases are correlated with high morbidity and mortality rates in humans. Previously, viral diseases such as influenza, dengue, HIV, and coronaviruses have resulted in epidemics or global pandemics, claiming many lives. Currently, the world is battling with coronavirus diseases 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) which has resulted in approximately 4 million deaths as of July 2021. The emergence of variants of this disease has also been challenging to the developed vaccines, and, as it stands, there are no effective therapeutic interventions against this disease. Proteins associated with viral infection can serve as molecular targets for disease prevention and treatment. Molecules involved in viral DNA replication and protein synthesis can be pivotal to these processes. Targets such as Papain-like protease (PLpro), 3-chymotrypsin like protease (3CLPro) also called main protease (Mpro), RNA-dependent RNA polymerase (RdRp), and helicase have been reported as potential points of treatment development for SARS-CoV-2 infection [1][2][3]. The SARS-CoV-2 spike protein and angiotensin-converting enzyme-2 (ACE-2) were also identified as promising targets for disease prevention [4][5][6]. The non-structural proteins essential for the replication of viral particles are specifically generated by PLpro and Mpro, defining their roles in viral replication and outlining their inhibition as potential anti-SARS-CoV-2 treatments [7][8][9].
Biologically active molecules have been screened against Mpro as either repurposed drugs or in the process of lead identification. Muhammad et al. [10] screened a library of phytochemicals against Mpro and revealed the potential usage of molecules from natural sources as anti-COVID-19 druggable candidates. In another study, bioactive medicinal plants were assayed against Mpro using in silico docking and pharmacological screening. Selected molecules from alkaloids and terpenoids were identified as inhibitors of this target, with a highly conserved inhibitory pattern to both SARS-CoV-2 and SARS-CoV [11]. Moreover, nucleopeptides and Opuntia-derived phytochemicals were suggested as Mpro potential inhibitors [12][13]. Therefore, the inhibition of protein activity of Mpro may potentially suppress coronavirus transmission.
Natural products of low molecular weight from plant sources are potent therapeutic agents for many diseases and some of these agents possess antiviral properties. While many natural products are fundamentally utilized as crude extracts, the purification of their active ingredients is essential for the prediction of their properties associated with the pharmacokinetics and pharmacodynamics of a drug molecule.
Accumulating evidence suggests that plant chemicals, for example, polyphenols and their functional derivatives such as flavonoids, saponins, and lignans can alter cellular functions, membrane permeability, and viral replication [14]. The role of phytochemicals has also been implicated in cell migration and proliferation, metabolism regulation (phytosterol, flavanols, anthocyanidins, cinnamic acids, etc.) [15][16], inflammatory processes (quercetin, kaempferol, etc.) [17], redox modulation (phenolics, curcumin, resveratrol, etc.) [18][19], and angiogenesis (astaxanthin) [20]. Ethanolic extract of Ficus benjamina has been shown to contain some active compounds such as rutin, kaempferol 3-O-rutinoside, and kaempferol 3-O-robinobioside, which were effective against herpes simplex [21]. Equally, the phytochemicals homolycorine and 2-O-acetyllycorine isolated from Leucojum vernum were shown to be effective against HIV-1 [22]. Rutin is a glycosylated flavonoid with a 3-rutinoside substitution. Its antiviral activity has been studied against avian influenza virus [23], herpes simplex [21], and parainfluenza-3 virus [24].
The world population is largely dependent on therapies from plant origin [25]. Compounds isolated from this source have little or no side effects with high biological specificity, chemical diversity, and targets multiple host sites by diverse pathways with negligible cost [26]. It was previously demonstrated that flavonoids, among other active compounds from plant sources, have been used for the treatment of HIV, herpes simplex, and influenza viruses, due to their antiviral properties [27]. These compounds inhibit viral replication, proteases, and reverse transcription [28]. Compounds such as quercetin, rutin, and myricetin have also been identified with similar properties [29].
2. ADME/Tox Prediction
The pharmacokinetics and toxicological properties of the ligands (Table 1 and Table 2) were analyzed according to previous methods to investigate how molecules can access the target site of Mpro after entering the bloodstream. This analysis is also crucial for analyzing the efficacy of molecules [30][31]. All parameters were within the ROF cut-off range for the test compounds and present no bystander toxicity effects since toxicity is the main task in developing new medications. Ames toxicity, carcinogenic properties, and rat acute toxicity were predicted in the current investigation.
Table 1. Properties of the selected compounds.
| Compound | ID a | M.W b | ROF c | QplogHERG d | QplogPoW e | QplogKP f | Donor HB | Acceptor HB | QplogS g | QplogBB h |
|---|---|---|---|---|---|---|---|---|---|---|
| Quercetin 3-O-Neohesperidoside | 5748416 | 610.5 | 2 | −6.449 | −1.998 | −6.423 | 9 | 20.55 | −2.932 | −4.728 |
| Myricetin 3-Rutinoside | 44259428 | 626.5 | 2 | −6.394 | −2.455 | −5.583 | 10 | 21.3 | −2.341 | −4.306 |
| Quercetin 3-Rhamnoside | 5353915 | 448.3 | 2 | −5.451 | −0.55 | −6.101 | 6 | 12.05 | −3.196 | −3.312 |
| Rutin | 5280805 | 610.5 | 2 | −5.238 | −2.495 | −7.251 | 9 | 20.55 | −2.175 | −4.503 |
| Myricitrin | 5281673 | 464.3 | 2 | −5.463 | −1.045 | −6.589 | 7 | 12.8 | −2.779 | −3.48 |
a Compound ID from PubChem database. b Formula weight of the compounds (acceptable range: 130.0–725 g/mol). c Number of permissible violations of Lipinski’s rule of five (acceptable range: maximum is 4). d Predicted IC50 value for blockage of HERG K+ channels (concern below −5.0). e Predicted octanol/water partition coefficient log p (acceptable range: −2.0 to 6.5). f Predicted skin permeability, log Kp (acceptable range: −8.0 to −1.0). g Predicted aqueous solubility; S in mol/L (acceptable range: −6.5 to 0.5). h Predicted brain/blood partition coefficient (acceptable range: −3.0 to 1.2). Donor HB (≤10); Acceptor HB (≤5).
Table 2. Toxicity analysis of the selected compounds predicted by AdmetSAR.
| Compound | ID | Ames Toxicity | Carcinogens | Acute Oral Toxicity | Rat Acute Toxicity |
|---|---|---|---|---|---|
| Quercetin 3-O-Neohesperidoside | 5748416 | AT | NC | III | 2.2619 |
| Myricetin 3-Rutinoside | 44259428 | NAT | NC | III | 2.4984 |
| Quercetin 3-Rhamnoside | 5353915 | NAT | NC | III | 2.5458 |
| Rutin | 5280805 | NAT | NC | III | 2.4984 |
| Myricitrin | 5281673 | NAT | NC | III | 2.5458 |
Note: AT: Ames toxic NAT: Non Ames toxic; NC: Non-carcinogenic; Category-III means (500 mg/kg > LD50 < 5000 mg/kg).
3. Docking Calculations
The protein-ligand interactions for all the complexes after the docking procedure were produced by Proteins Plus at https://proteins.plus/ (accessed on 1 August 2021) as depicted in Figure 1. The model representation of the best pose against decoy poses was also presented using Mpro-Quercetin-3-O-Neohesperidoside (Figure 2). The binding properties, such as the scoring functions of Autodock Vina and MM/GBSA, the number of hydrogen bond integrations, and types of residues and their distances (Å) are tabulated in Table 3. In addition, the possible residue interaction crucial to the inhibition process of Mpro by these flavonoids was proposed in Figure 3.
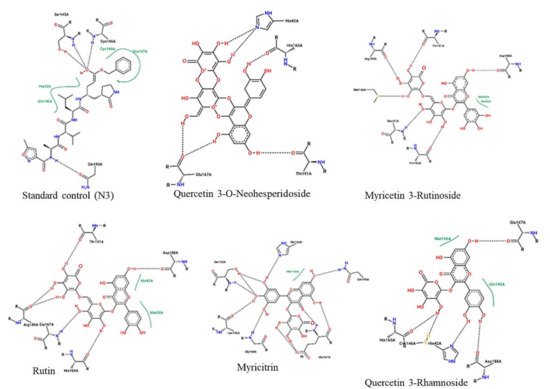
Figure 1. 2-dimentional protein–ligand interactions created for docked ligands and N3 into the active site of Mpro ranked according to their binding energies. Black bond interactions showed H-bonds between the atoms of the ligands and the residues of the receptor.
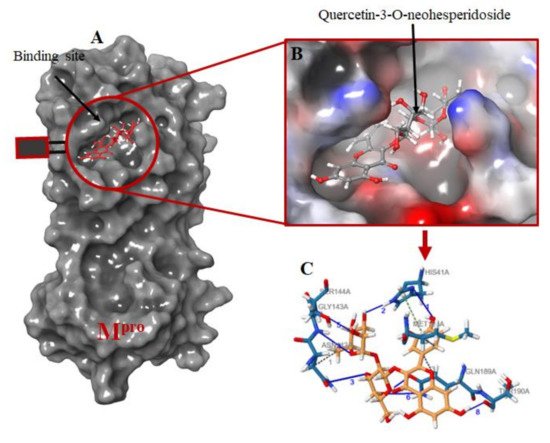
Figure 2. Molecular docking of the ligands into Mpro binding site. (A) the docking of the selected ligands in Mpro binding pocket; (B) Quercetin-3-O-Neohesperidoside seats perfectly in the Mpro binding pocket; (C) interacting atoms of Quercetin-3-O-Neohesperidoside and Mpro residues in the binding pocket. Note: blue lines indicate hydrogen bond interaction; green dotted lines indicate pi-stacking, while gray dotted lines depict hydrophobic interactions.
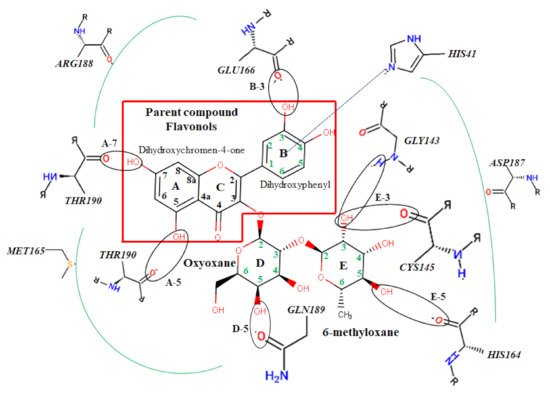
Figure 3. Atomic interaction of flavonoids with Mpro residues. While several residues form hydrophobic interactions with the ligands, other interactions such as hydrogen, π-cation, and π-π stacking were also involved in the inhibition mechanism of Mpro.
Table 3. Binding energies of flavonoids docked against Mpro.
| Name | Dock Score | ∆G Bind | H-Bond | Residues (Å) | Other Bond (Å) |
|---|---|---|---|---|---|
| Standard | −16.5 | −80.88 | 6 | CYS145 (2.13), LEU141 (2.76), PHE140 (2.02), GLU166 (1.68,1.83, 2.05) | Salt bridges (2) |
| Quercetin 3-O-Neohesperidoside | −16.8 | −87.60 | 5 | GLY143 (2.76), CYS145 (2.11), GLN189 (2.11), THR190 (1.76), HIS41 (2.30) | π-π stacking HIS41 (1.49) |
| Myricetin 3-Rutinoside | −12.9 | −87.50 | 7 | CYS145 (2.08), ASN142 (1.75), GLU166 (1.98), THR190 (2.21), ARG188 (1.97), HIS164 (1.90,1.98) | |
| Quercetin 3-Rhamnoside | −10.3 | −80.17 | 4 | LEU141 (1.49), THR190 (1.78), GLU166 (2.01), HIS164 (1.81) | |
| Rutin | −10.0 | −58.95 | 6 | THR190 (1.83), HIS41 (2.08), GLY143 (2.39,1.89), ASN142 (1.92), LEU141 (2.10) | - |
| Myricitrin | −9.1 | −49.22 | 3 | CYS145 (2.51), ASN142 (2.04), THR190 (1.83) | π-π stacking HIS41 (5.37) |
4. Molecular Dynamic Simulation
To associate structural and mechanistic information with experimental data, the MDs were carried out. The Mpro-ligand complexes were computationally simulated for 100 ns to decipher the complex stability and dynamic behavior as presented in Figure 4, Figure 5 and Figure 6, and Table 2. The root-mean-square deviation (RMSD) of the five ligands were plotted against 1000 frame indexes for 100 ns (Figure 4a). The root-mean-square fluctuation (RMSF) of the C-alpha of the protein complexed with these ligands was also plotted against residues (Figure 4b). The ligand properties were also taken into account by plotting the radius of gyration (rGyr) for all five complexes against the frame index over the 100 ns simulation time (Figure 5). The simulation properties were calculated as the mean ± SD for RMSD, RMSF, and rGyr (Table 4). Finally, protein interactions with the ligands were monitored throughout the simulation. These interactions were categorized by type and summarized as shown in Figure 6.
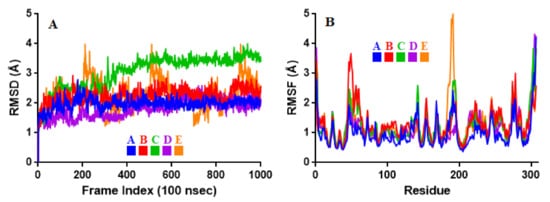
Figure 4. Stability and flexibility of the selected flavonoid derivatives complexed with Mpro over the course of 100 ns. (A) is the RMS deviation and (B) is the RMS functions of the Cα of each complexes. Color codes denote: Quercetin-3-O-Neohesperidoside (A), Quercetin 3-Rhamnoside (B), Myricitrin (C), Rutin (D), and Myricetin 3-Rutinoside (E).
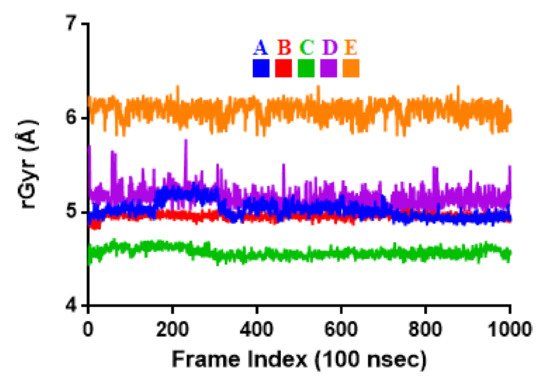
Figure 5. Radius of gyration (rGyr) plot for Mpro and the selected flavonoid derivatives within the simulation time of 100 ns. Color codes denote: Quercetin-3-O-Neohesperidoside (A), Quercetin 3-Rhamnoside (B), Myricitrin (C), Rutin (D), and Myricetin 3-Rutinoside (E).
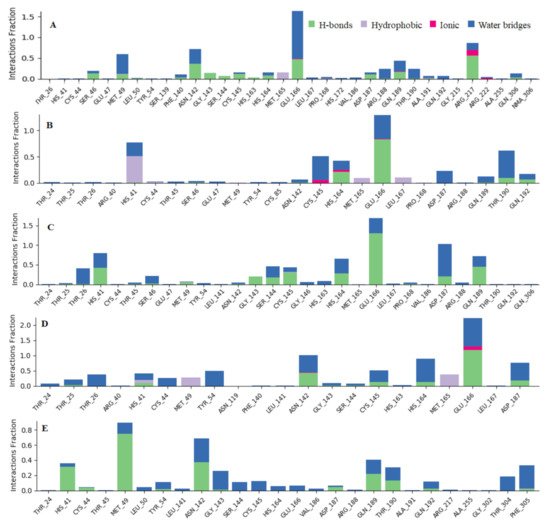
Figure 6. Observed Mpro-ligands interaction during the 100 ns MD simulation. Interactions include; hydrogen bonds, hydrophobic, ionic and water bridges. Letter codes indicate: Quercetin-3-O-Neohesperidoside (A), Quercetin 3-Rhamnoside (B), Myricitrin (C), Rutin (D), and Myricetin 3-Rutinoside (E).
Table 4. The simulation properties of the complexes.
| Properties | A | B | C | D | E |
|---|---|---|---|---|---|
| RMSD | 1.98 ± 0.19 | 2.25 ± 0.26 | 3.05 ± 0.57 | 1.81 ± 0.30 | 2.26 ± 0.51 |
| RMSF | 1.00 ± 0.51 | 1.31 ± 0.55 | 1.27 ± 0.58 | 1.15 ± 0.58 | 1.25 ± 0.65 |
| rGyr | 5.03 ± 0.09 | 4.96 ± 0.03 | 4.57 ± 0.05 | 5.15 ± 0.10 | 6.09 ± 0.09 |
Values represent the mean ± SD of 1000 frame index replicates. Letter codes: Quercetin-3-O-Neohesperidoside (A), Quercetin 3-Rhamnoside (B), Myricitrin (C), Rutin (D), and Myricetin 3-Rutinoside (E). Values are in Å.
References
- Muhammed, Y. Molecular targets for COVID-19 drug development: Enlightening Nigerians about the pandemic and future treatment. Biosaf. Health 2020, 2, 210–216.
- Yuan, Y.; Cao, D.; Zhang, Y.; Ma, J.; Qi, J.; Wang, Q.; Lu, G.; Wu, Y.; Yan, J.; Shi, Y. Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat. Commun. 2017, 8, 15092.
- Dong, S.; Sun, J.; Mao, Z.; Wang, L.; Lu, Y.; Li, J. A guideline for homology modeling of the proteins from newly discovered betacoronavirus, 2019 novel coronavirus (2019-nCoV). J. Med. Virol. 2020, 92, 1542–1548.
- Liu, C.; Zhou, Q.; Li, Y.; Garner, L.V.; Watkins, S.P.; Carter, L.J.; Smoot, J.; Gregg, A.C.; Daniels, A.D.; Jervey, S. Research and Development on Therapeutic Agents and Vaccines for Covid-19 and Related Human Coronavirus Diseases; ACS Publications: Washington, DC, USA, 2020.
- Schoeman, D.; Fielding, B.C. Coronavirus envelope protein: Current knowledge. Virol. J. 2019, 16, 69.
- Fadaka, A.O.; Sibuyi, N.R.S.; Adewale, F.; Bakare, O.O.; Akanbi, M.O.; Klein, A.; Madiehe, A.M.; Meyer, M. Understanding the epidemiology, pathophysiology, diagnosis and management of SARS-CoV-2. J. Int. Med. Res. 2020, 48, 0300060520949077.
- Needle, D.; Lountos, G.T.; Waugh, D.S. Structures of theMiddle East respiratory syndrome coronavirus3C-like protease reveal insights into substrate specificity. Acta Crystallogr. Sect. D Biol. Crystallogr. 2015, 71, 1102–1111.
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus Main Proteinase (3CLpro) Structure: Basis for Design of Anti-SARS Drugs. Science 2003, 300, 1763–1767.
- Mody, V.; Ho, J.; Wills, S.; Mawri, A.; Lawson, L.; Ebert, M.C.C.J.C.; Fortin, G.M.; Rayalam, S.; Taval, S. Identification of 3-chymotrypsin like protease (3CLPro) inhibitors as potential anti-SARS-CoV-2 agents. Commun. Biol. 2021, 4, 93.
- Qamar, M.T.U.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.-L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020, 10, 313–319.
- Gyebi, G.A.; Ogunro, O.B.; Adegunloye, A.P.; Ogunyemi, O.M.; Afolabi, S.O. Potential inhibitors of coronavirus 3-chymotrypsin-like protease (3CLpro): An in silico screening of alkaloids and terpenoids from African medicinal plants. J. Biomol. Struct. Dyn. 2020, 39, 3396–3408.
- Roviello, V.; Musumeci, D.; Mokhir, A.; Roviello, G.N. Evidence of protein binding by a nucleopeptide based on a thymine-decorated L-diaminopropanoic acid through CD and in silico studies. Curr. Med. Chem. 2021, 28, 1.
- Vicidomini, C.; Roviello, V.; Roviello, G.N. In Silico Investigation on the Interaction of Chiral Phytochemicals from Opuntia ficusindica with SARS-CoV-2 Mpro. Symmetry 2021, 13, 1041.
- Upadhyay, S.; Dixit, M. Role of Polyphenols and Other Phytochemicals on Molecular Signaling. Oxid. Med. Cell. Longev. 2015, 2015, 504253.
- Graf, B.L.; Raskin, I.; Cefalu, W.T.; Ribnicky, D.M. Plant-derived therapeutics for the treatment of metabolic syndrome. Curr. Opin. Investig. Drugs 2010, 11, 1107.
- García-Lafuente, A.; Guillamón, E.; Villares, A.; Rostagno, M.A.; Martínez, J.A. Flavonoids as an-ti-inflammatory agents: Implications in cancer and cardiovascular disease. Inflamm. Res. 2009, 58, 537–552.
- Ichikawa, D.; Matsui, A.; Imai, M.; Sonoda, Y.; Kasahara, T. Effect of various catechins on the IL-12p40 pro-duction by murine peritoneal macrophages and a macrophage cell line, J774. 1. Biol. Pharm. Bull. 2004, 27, 1353–1358.
- Loo, G. Redox-sensitive mechanisms of phytochemical-mediated inhibition of cancer cell proliferation (review). J. Nutr. Biochem. 2003, 14, 64–73.
- Tosetti, F.; Noonan, D.M.; Albini, A. Metabolic regulation and redox activity as mechanisms for angiopre-vention by dietary phytochemicals. Int. J. Cancer 2009, 125, 1997–2003.
- Kowshik, J.; Baba, A.B.; Giri, H.; Reddy, G.D.; Dixit, M.; Nagini, S. Astaxanthin Inhibits JAK/STAT-3 Signaling to Abrogate Cell Proliferation, Invasion and Angiogenesis in a Hamster Model of Oral Cancer. PLoS ONE 2014, 9, e109114.
- Yarmolinsky, L.; Huleihel, M.; Zaccai, M.; Ben-Shabat, S. Potent antiviral flavone glycosides from Ficus benjamina leaves. Fitoterapia 2012, 83, 362–367.
- Szlávik, L.; Gyuris, Á.; Minárovits, J.; Forgo, P.; Molnár, J.; Hohmann, J. Alkaloids from Leucojum vernum and antiretroviral activity of Amaryllidaceae alkaloids. Planta Med. Nat. Prod. Med. Plant Res. 2004, 70, 871–873.
- Ibrahim, A.K.; Youssef, A.I.; Arafa, A.; Ahmed, S.A. Anti-H5N1 virus flavonoids from Capparis sinaica Veill. Nat. Prod. Res. 2013, 27, 2149–2153.
- Orhan, D.D.; Özçelik, B.; Özgen, S.; Ergun, F. Antibacterial, antifungal, and antiviral activities of some flavonoids. Microbiol. Res. 2010, 165, 496–504.
- Ekor, M. The growing use of herbal medicines: Issues relating to adverse reactions and challenges in monitoring safety. Front. Pharmacol. 2014, 4, 177.
- Lin, L.-T.; Hsu, W.-C.; Lin, C.-C. Antiviral Natural Products and Herbal Medicines. J. Tradit. Complement. Med. 2014, 4, 24–35.
- Pour, P.M.; Fakhri, S.; Asgary, S.; Farzaei, M.H.; Echeverria, J. The signaling pathways, and ther-apeutic targets of antiviral agents: Focusing on the antiviral approaches and clinical perspectives of antho-cyanins in the management of viral diseases. Front. Pharmacol. 2019, 10, 1207.
- Kurapati, K.R.V.; Atluri, V.S.; Samikkannu, T.; Garcia, G.; Nair, M.P.N. Natural Products as Anti-HIV Agents and Role in HIV-Associated Neurocognitive Disorders (HAND): A Brief Overview. Front. Microbiol. 2016, 6, 1444.
- Ninfali, P.; Antonelli, A.; Magnani, M.; Scarpa, E.S. Antiviral Properties of Flavonoids and Delivery Strategies. Nutrition 2020, 12, 2534.
- Ojo, O.A.; Aruleba, R.T.; Adekiya, T.A.; Sibuyi, N.R.S.; Ojo, A.B.; Ajiboye, B.O.; Oyinloye, B.E.; Adeola, H.A.; Fadaka, A.O. Deciphering the interaction of puerarin with cancer macromolecules: An in silico investigation. J. Biomol. Struct. Dyn. 2020, 1–12.
- Fadaka, A.O.; Aruleba, R.T.; Sibuyi, N.R.S.; Klein, A.; Madiehe, A.M.; Meyer, M. Inhibitory potential of re-purposed drugs against the SARS-CoV-2 main protease: A computational-aided approach. J. Biomol. Struct. Dyn. 2020.
More
Information
Subjects:
Pathology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Entry Collection:
COVID-19
Revisions:
2 times
(View History)
Update Date:
10 Sep 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No