 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Lukas Junk | + 3349 word(s) | 3349 | 2021-08-13 11:19:10 | | | |
| 2 | Conner Chen | Meta information modification | 3349 | 2021-09-03 09:56:14 | | |
Video Upload Options
Ilamycins/rufomycins and cyclomarins are marine cycloheptapeptides containing unusual amino acids. Produced by Streptomyces sp., these compounds show potent activity against a range of mycobacteria, including multidrug-resistant strains of Mycobacterium tuberculosis. The cyclomarins are also very potent inhibitors of Plasmodium falciparum. Biosynthetically the cyclopeptides are obtained via a heptamodular nonribosomal peptide synthetase (NRPS) that directly incorporates some of the nonproteinogenic amino acids. A wide range of derivatives can be obtained by fermentation, while bioengineering also allows the mutasynthesis of derivatives, especially cyclomarins. Other derivatives are accessible by semisynthesis or total synthesis, reported for both natural product classes.
1. Discovery of Anti-Tubercular Cycloheptapeptides
1.1. Discovery of the Ilamycins/Rufomycins
In 1962, two independent research groups investigated marine Streptomycetes from soil samples found on Japanese islands. Takita et al. observed that the culture filtrate of a new strain, Streptomyces insulates (A-165-Z1), later renamed Streptomyces islandicus, inhibited the growth of Mycobacterium 607 and Mycobacterium phlei. They isolated two antibiotics and named them ilamycin A and B (IlaA and IlaB)[1][2]. In addition, at this time Shibata et al. isolated two new antibiotics, rufomycin I and II (Ruf I and Ruf II), from the newly discovered Streptomyces atratus (46408), found to be especially active against acid-fast bacteria[3][4]. The compounds were also active against Mycobacterium tuberculosis and Mycobacterium smegmatis but almost inactive against most other bacteria, fungi, and yeasts. Subsequent research indicated that these two antibiotics possess very similar chemical structures [5][6][7][8][9][10].
The structures of ilamycins/rufomycins (Figure 1) are unusual, as these cyclic heptapeptides contain a series of atypical amino acids. Most prominent is the N-prenylated tryptophan ①, which can also be found in the epoxidized form [11][12]. At the N-terminus of the tryptophan, a γ,δ-unsaturated amino acid is incorporated ⑦ [13]. Common to all derivatives is a unique 3-nitrotyrosine ③, a building block not found in any other natural product. The greatest variability is observed in the leucine building block ⑤, which can be oxidized to different oxidation levels at a terminal methyl group. In its original description, ilamycin was proposed to contain an aldehyde functionality[10], but structural elucidation by NMR and X-ray crystallography showed that the aldehyde functionality undergoes cyclization with the nearby amide bond [14][15][16]. Very recently, a wide range of new ilamycins/rufomycins were described, differing mainly in the combination of different amino acid oxidation levels ⑤ and the N-prenyl substituent of ① (alkene, epoxide, diol)[14][15][16].
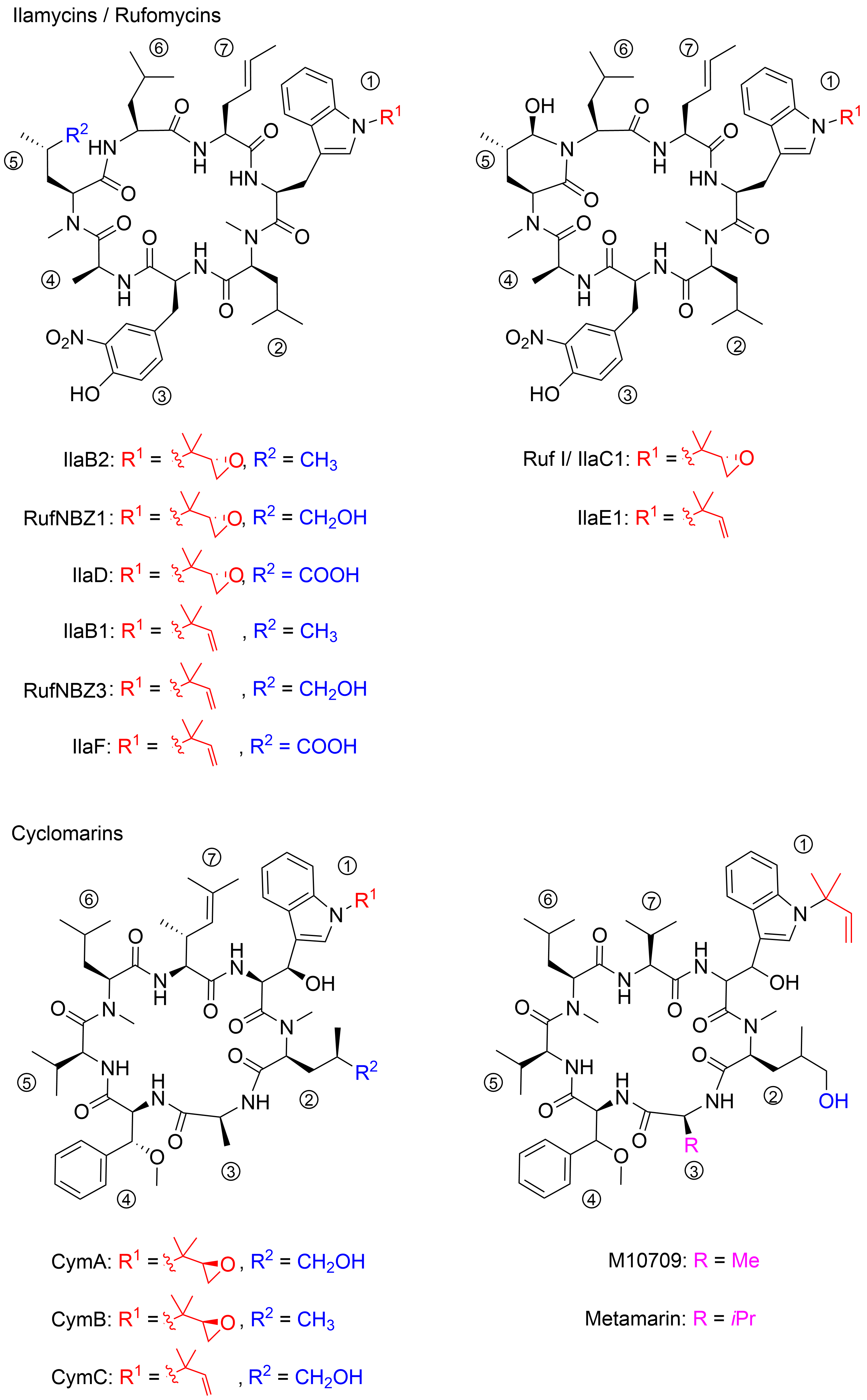
Figure 1. Selected ilamycins/rufomycins and cyclomarins.
1.2. Discovery of the Cyclomarins
In 1999, the research groups of Fenical and Clardy reported the isolation of three new anti-inflammatory cyclic peptides from extracts of a Streptomyces sp. collected in Mission Bay, California[17]. These secondary metabolites from the strain CNB-982, called cyclomarins (Cym) A−C, are structurally related to the rufomycins. Very similar amino acid building blocks are incorporated, although in a different sequence. As in the rufomycins, an N-prenylated tryptophan ①(CymC) is a notable building block that can also be epoxidized (CymA). However, in contrast to the rufomycins, in the cyclomarin series, the tryptophan units are β-hydroxylated. At the N-terminus of the tryptophan, a γ,δ-unsaturated amino acid is incorporated, not a linear one as found in the rufomycins, but one that is branched and dimethylated ⑦. One of the leucines is also oxidized at the δ position ②, at least in CymA and C, but at another position, as in the rufomycins. Most obvious is the replacement of the unique nitrotyrosine by another aromatic amino acid, syn-β-methoxyphenylalanine ④.
In 2010, the group of Mikami described the extraction of a new cyclomarin derivative M10709 from clinically isolated Streptomyces sp. IFM 10,709 [18]. Although not all stereogenic centers were determined properly, results revealed the compound was different from cyclomarin C only by the replacement of the unsaturated amino acid ⑦ by valine. The same structural motif was also found in a recently isolated metamarine in which a valine at position ③ replaced an alanine. The discovery of metamarine resulted from a larger soil metagenome project undertaken to discover rufomycin/cyclomarin-like antibiotics[19].
2. Total Syntheses of Marine Cycloheptapeptides
The interesting biological properties and unusual building blocks of marine cycloheptapeptides sparked the interest of synthetic chemists, and the syntheses of several different amino acids and fragments have been reported in a recent review [20]. Therefore, they will not be discussed in detail here, and the focus will be on the total syntheses of the natural products.
2.1. Total Synthesis of Ilamycins/Rufomycins
To date, only one synthetic route has been described for ilamycins E1 and F by Guo and Ye et al.[21] In their highly convergent strategy, the ilamycins were synthesized from two parts (1 and 2) that were linked between ① and ⑦ to the macrocyclic lactam (Scheme 1). The lower right tripeptide part 1 (①–③) was prepared in five steps from tryptophan, while the upper left tetrapeptide 2 (④–⑦) required 13 steps from glutamic acid. Final oxidation of the δ-hydroxyleucine ⑤ resulted in the described ilamycins.
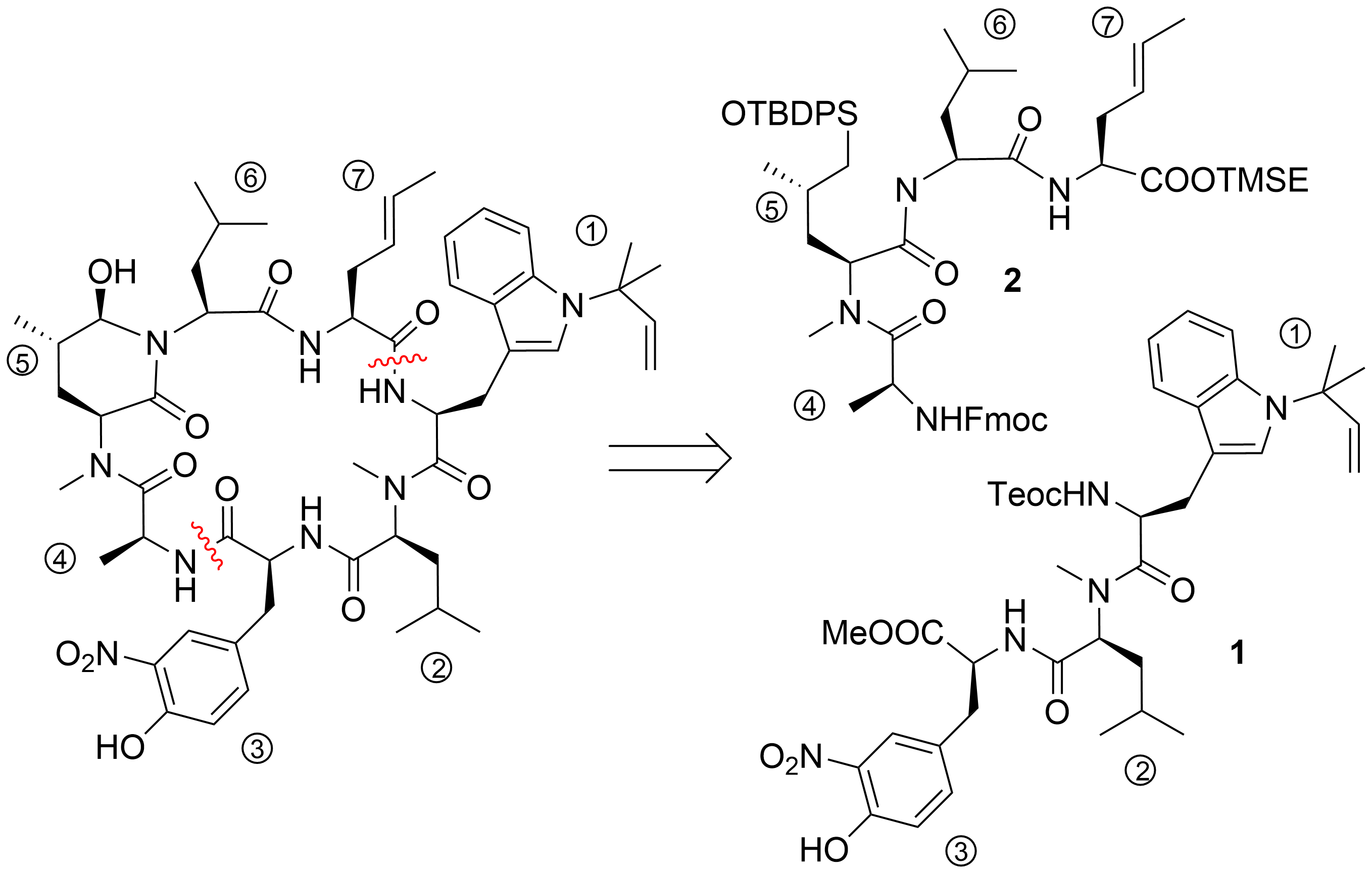
Scheme 1. Retrosynthesis of ilamycins (Guo and Ye).
The synthesis of peptide fragment 1 was rather straightforward (Scheme 2). 2-(Trimethylsilyl)ethoxycarbonyl (Teoc)-protected tryptophan methylester 3 was subjected to a Pd-catalyzed N-tert-prenylation according to a protocol developed by Baran et al. [22]. Saponification of the ester moiety of 4 and peptide coupling with N-methylated Leu-OMe produced dipeptide 5, which was further elongated to tripeptide 1.
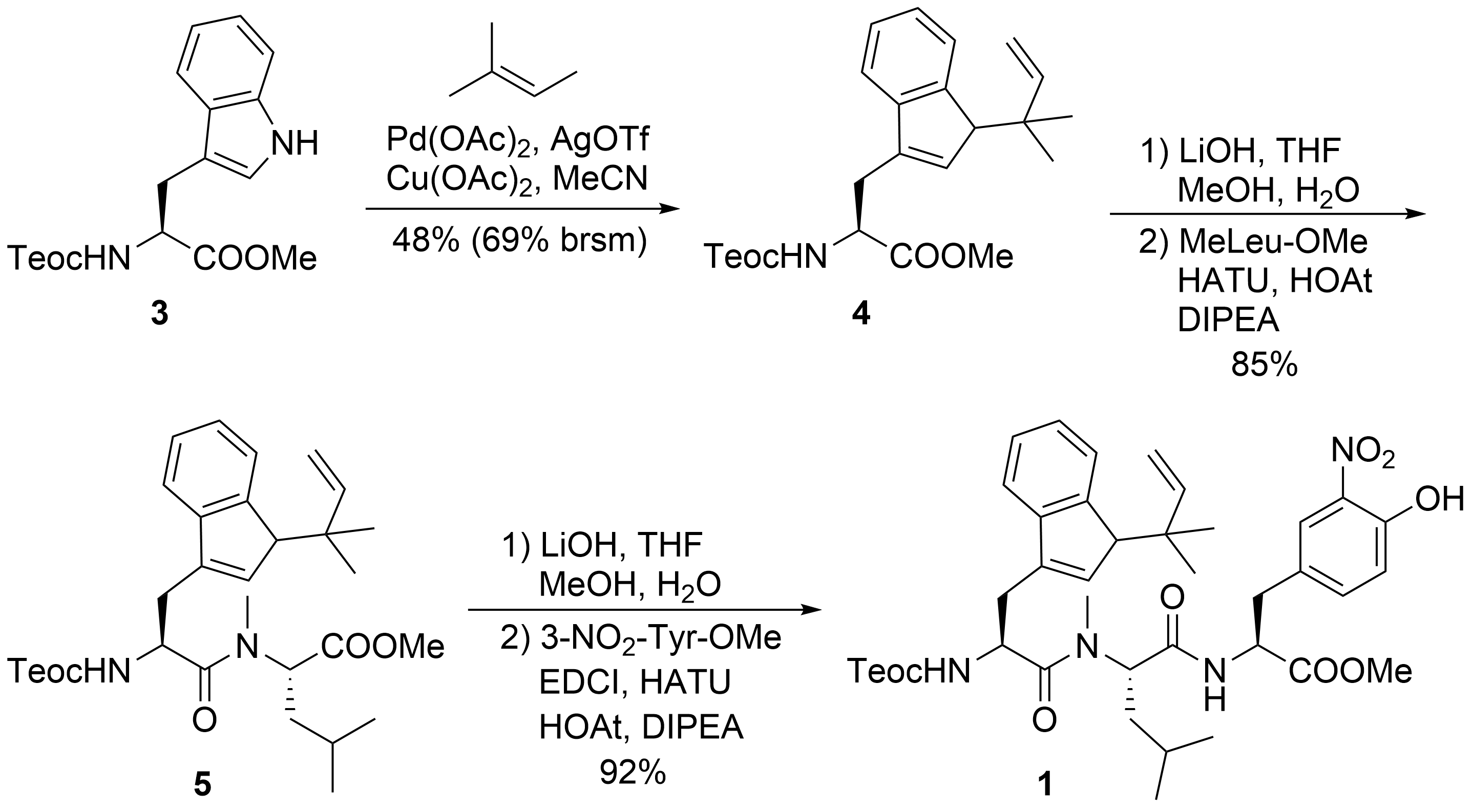
Scheme 2. Synthesis of peptide fragment 1.
For the larger fragment 2, glutamic acid was converted into protected 8 (Scheme 3) according to a synthetic route developed during the synthesis of dysithiazolamide [23]. The glutamic acid was converted into the dimethyl ester and N-Boc protected before it was stereoselectively α-methylated at the sterically least-hindered ester moiety [24][25]. For the chemoselective reduction of the γ-ester 6, a second N-Boc-protecting group was introduced, and the sterically least-hindered ester functionality was reduced with DIBAL-H. Silyl protection of the primary alcohol and subsequent mono-Boc deprotection yielded 7. The methyl ester was saponified (to avoid α-methylation), and the Boc-amide was selectively N-methylated to 8 with NaH/MeI. The free carboxylic acid 8 was converted into the corresponding benzyl ester. TMSOTf/NEt3 was used for selective cleavage of the N-Boc-protecting group without affecting the OTBDPS group. The free amine could be coupled with Fmoc-protected alanine, and the C-terminal benzyl ester was cleaved by catalytic hydrogenation to provide the free acid 9.
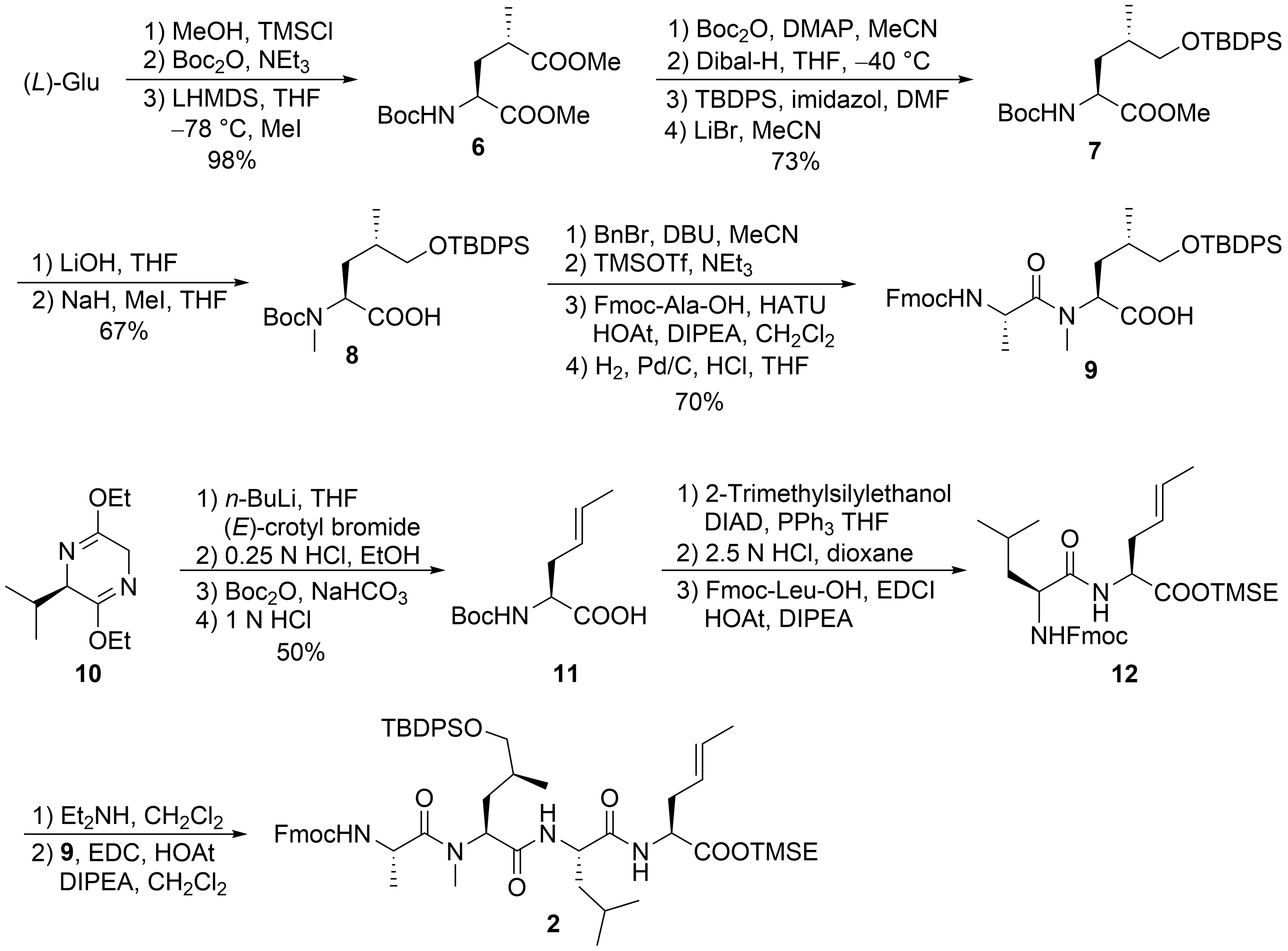
Scheme 3. Synthesis of peptide fragment 2.
Schöllkopf auxiliary 10[26] was subjected to stereoselective crotylation to generate the C-terminal unsaturated amino acid of 2 (Scheme 3). Subsequent auxiliary cleavage provided N-Boc-protected amino acid 11, which was converted into the corresponding TMSE ester. Boc-deprotection and peptide coupling produced dipeptide 12. Subsequent Fmoc deprotection and coupling with 9 generated the linear tetrapeptide 2.
With the two major building blocks produced, ilamycin synthesis could proceed to the final step (Scheme 4). Mild saponification of the methylester 1 and coupling with Fmoc-deprotected 2 using (2-azabenzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyAOP)[27] yielded 13 without significant epimerization. Global deprotection with TBAF resulted in the linear heptapeptide, which was subjected to macrolactamization. While many peptide coupling reagents have been investigated, the best results were obtained using pentafluorophenyl diphenylphosphinate (FDPP)1[28]. As a side product in addition to the expected cyclopeptide 14, the diphenylphosphinylated ester was formed, which could directly be converted into 14 by treatment with K2CO3 in methanol, providing an overall yield of 43% of the desired 14. Finally, only the primary OH-functionality needed to be oxidized. Depending on the oxidation protocol, both ilamycin E1 and F could be obtained. Ilamycin E1 was obtained as a single stereoisomer. Notably, ilamycin F is also available on a gram scale via fermentation, but the derivative E1, which is approximately 100-fold more potent, is not. Therefore, the authors developed a protocol to convert ilamycin F into intermediate 14 by reducing the mixed anhydride, permitting an interconversion of ilamycin F into ilamycin E1 [21].
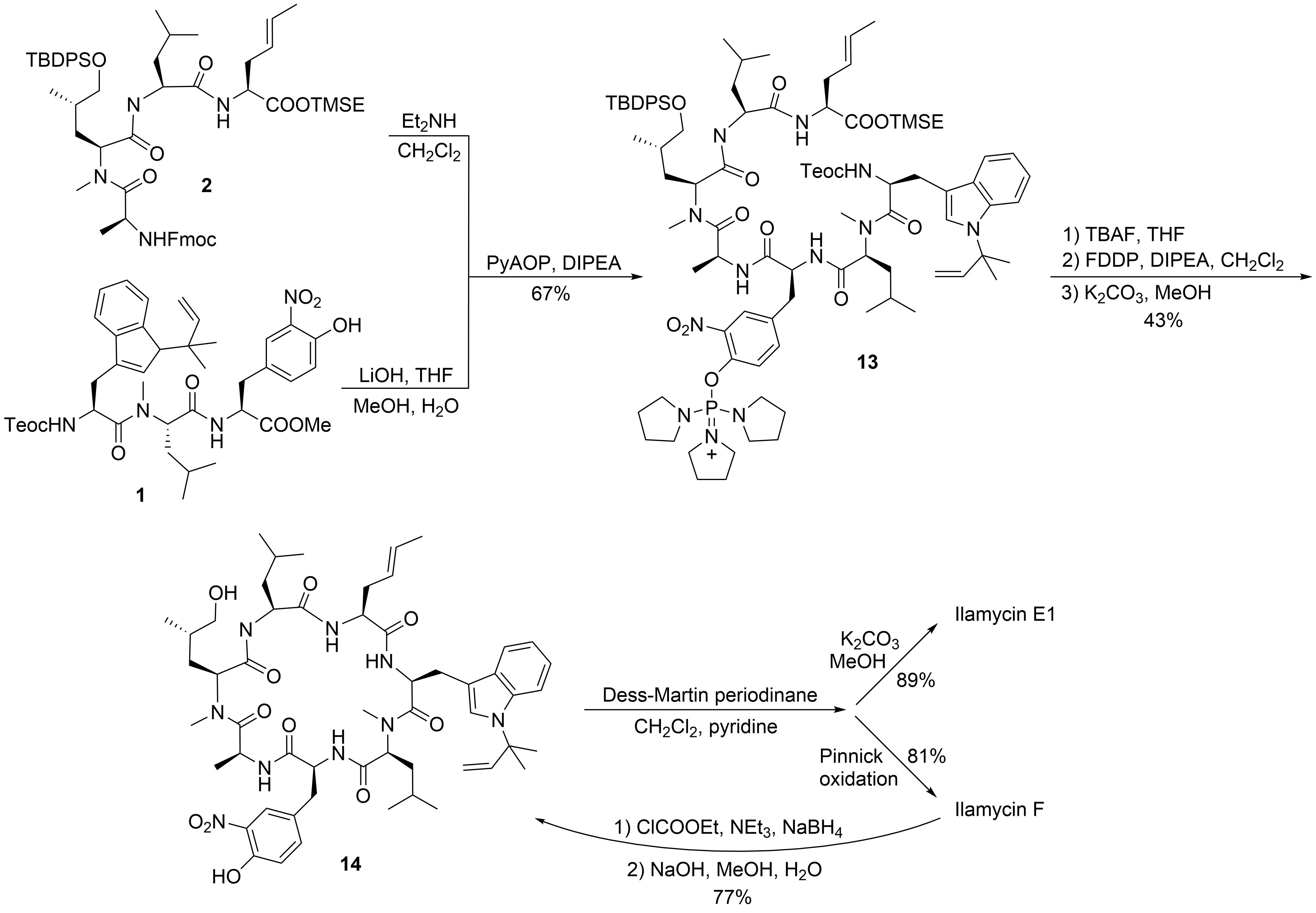
Scheme 4. Synthesis of ilamycinE1 and F.
2.2. Total Synthesis of Cyclomarins
The amino acids present in the cyclomarins are slightly more complex than in the ilamycins, and various synthetic approaches have been undertaken to produce these building blocks and partial structures of cyclomarin [29]. These are covered in a recent review [20], and therefore only the routes applicable to the synthesis of cyclomarins and derivatives will be discussed here.
The first synthesis of cyclomarin C was reported in 2004 by Yao and coworkers [30]. The unusual tert-prenylated β-hydroxy-tryptophan ① was obtained from indole derivative 15 (Scheme 5). This compound is available from indole via N-alkylation with ethyl-α-bromo-propionate, subsequent α-methylation of the ester, LAH-reduction, and acetylation [31]. Formylation and a subsequent Horner–Wadsworth–Emmons reaction yielded α,β-unsaturated ester 16, which could be subjected to a Sharpless aminohydroxylation [32]. Moderate yield and enantioselectivity of the desired β-hydroxytryptophan derivative 17 was obtained. Unfortunately, no comment was made concerning the regioselectivity of the reaction. Silylation of the β-hydroxy group and selective transesterification of the acetate gave rise to primary alcohol 18, which could be oxidized to the aldehyde and methenylated via Wittig reaction. Finally, the Cbz-protecting group from 19 was removed selectively without affecting the generated double bond. Furthermore, the free amine was Fmoc-protected after saponification of the ester. The use of the Fmoc- or Alloc-protecting group is essential for the synthesis of cyclomarins because other protecting groups, such as Boc, cannot be removed later on without side reactions, such as the elimination of the β-hydroxy functionality[33].
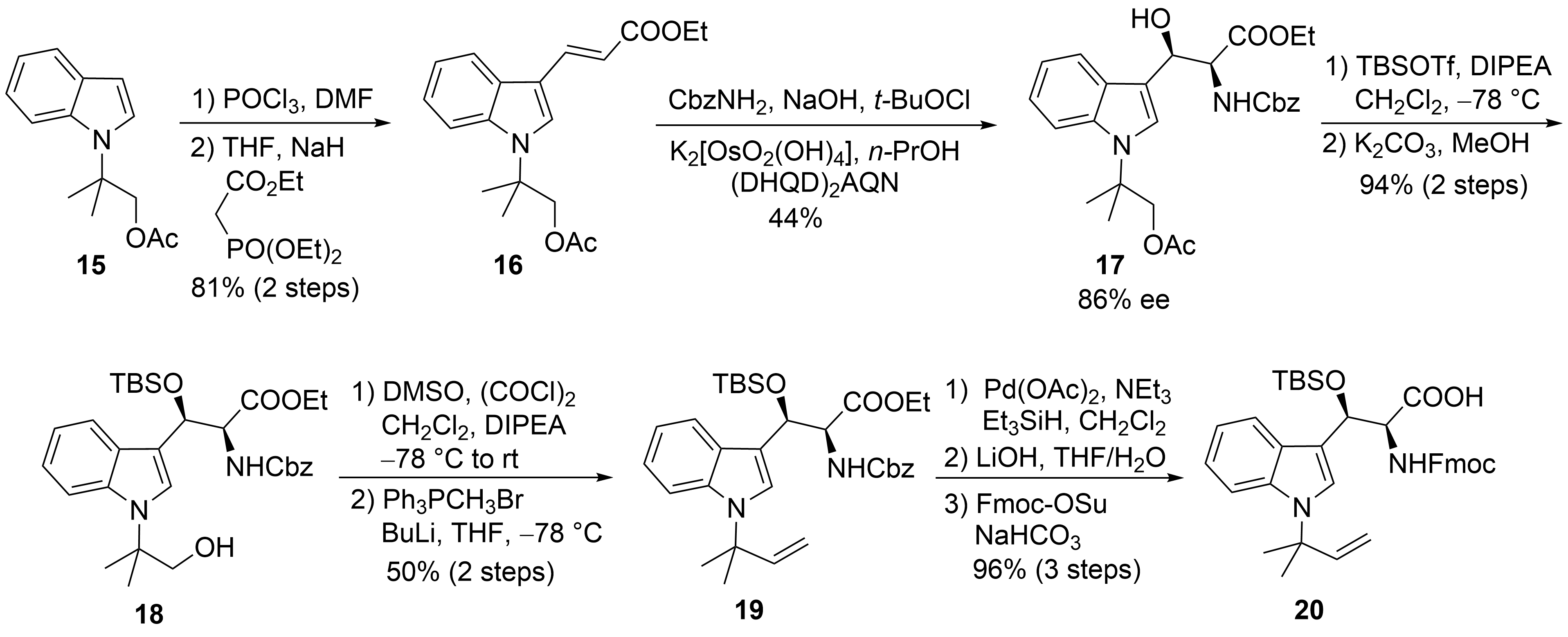
Scheme 5. Synthesis of protected tryptophan 20 (building block ①).
The synthesis of δ-hydroxyleucine building block ②, with the opposite configuration of the γ-methyl group than in amino acid ⑤ in the ilamycins, was obtained by classical asymmetric synthesis using chiral auxiliary chemistry (Scheme 6). According to Evans et al.[34], chiral oxazolidinone 21 was subjected as its titanium enolate in a Michael addition to tert-butyl acrylate to provide a good yield of 22 with high stereoselectivity. The imide was selectively reduced in the presence of the tert-butyl ester using LiBH4, and the resulting primary alcohol was O-benzoylated to 23. Acidic cleavage of the tert-butyl ester, activation of the carboxylic acid, and reapplication of the Evans auxiliary A provided oxazolidinone 24. Deprotonation of 24 and stereoselective azidation produced azide 25. Catalytic hydrogenation in the presence of Boc2O resulted in the formation of the N-Boc-protected amino derivative, which was saponified to the corresponding amino acid 26. The desired N-methyl group was introduced by conversion of 26 into the corresponding oxazolidinone 27, which was reduced using triethylsilane in presence of trifluoroacetic acid. Subsequent cleavage of the Boc-protecting group required its reintroduction to 28.
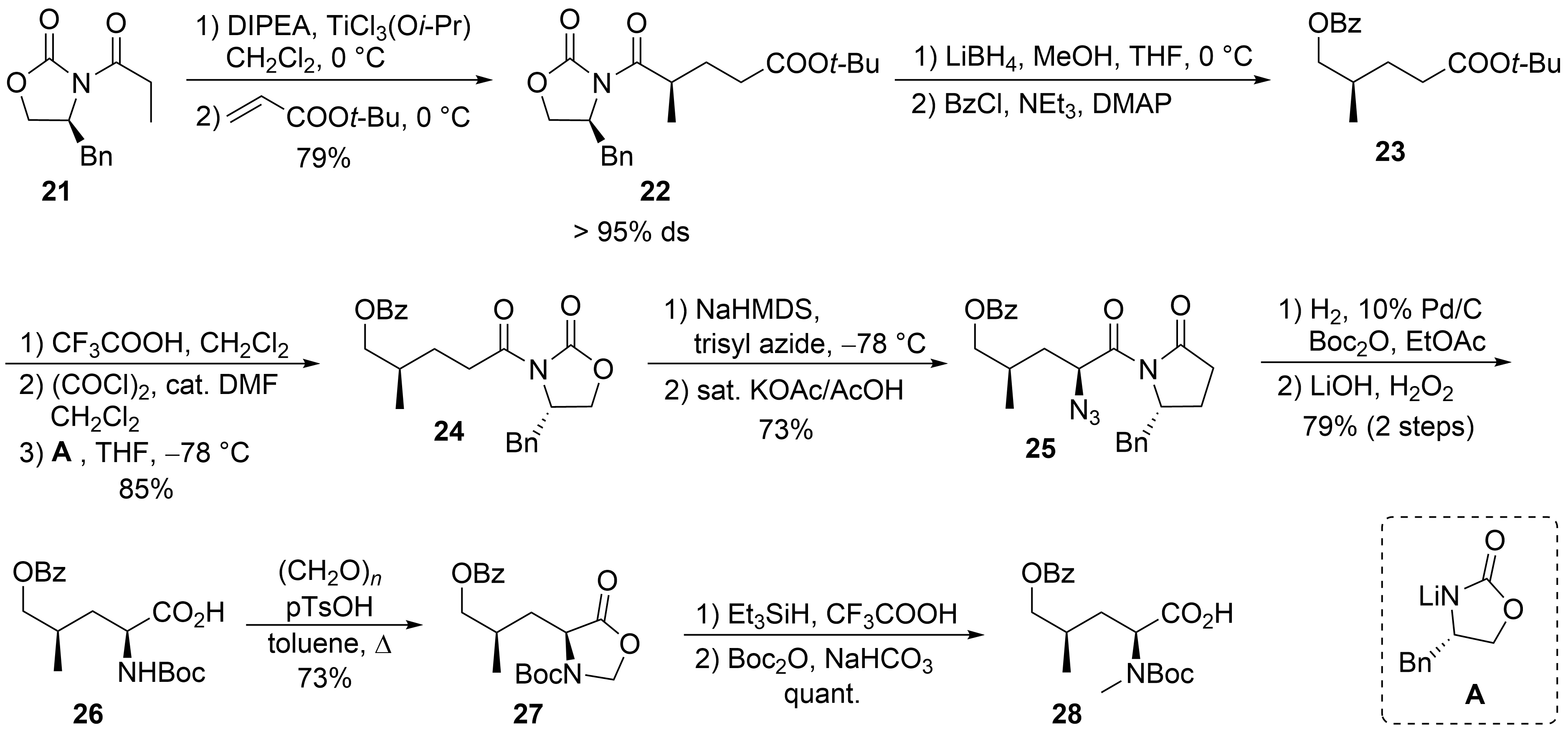
Scheme 6. Synthesis of protected hydroxyleucine 28 (building block ②).
The unusual β-methoxyphenylalanine ④ was obtained from N-phthaloyl-protected phenylalanine 29, which was converted into the corresponding tert-butylamide 30 (Scheme 7). Oxygen functionality was introduced into the β-position by subjecting 30 to a Wohl–Ziegler bromination, providing a 1:1 diastereomeric mixture of the desired β-bromo derivative[35]. According to Easton et al., treatment of the diastereomeric mixture with AgNO3 in aqueous acetone produced the desired (2S,3R)-β-hydroxyphenylalanine enantio- and diastereoselectively[36]. The stereochemical outcome can be explained by a preferred conformation of the benzylic carbenium ion in the substitution step. The best selectivities were obtained with the tert-butylamide. Subsequent O-methylation provided the methoxy derivative 31, converted into the N-Boc-protected amino acid 32 under standard conditions.

Scheme 7. Synthesis of protected β-methoxy phenylalanine 32 (building block ④).
Finally, the unsaturated amino acid ⑦ was obtained via an asymmetric chelate enolate Claisen rearrangement, developed by Kazmaier et al. (Scheme 8)[37][38]. Trifluoroacetyl (TFA)-protected glycine crotyl ester 33 was deprotonated and converted into a chelated aluminum ester enolate, which in the presence of quinidine underwent a [3,3]-sigmatropic rearrangement to unsaturated amino acid 34 with good yield and enantioselectivity. Epimerization of the α-stereogenic center was avoided by first converting 34 into the Boc-protected ester 35 and then, in a second step, into the corresponding phthaloyl-protected derivative 36. A direct epimerization-free conversion (34 to 36) was not possible. Ozonolysis of the double bond and subsequent Wittig reaction produced protected amino acid 37, finally converted into the Fmoc-protected acid 38.
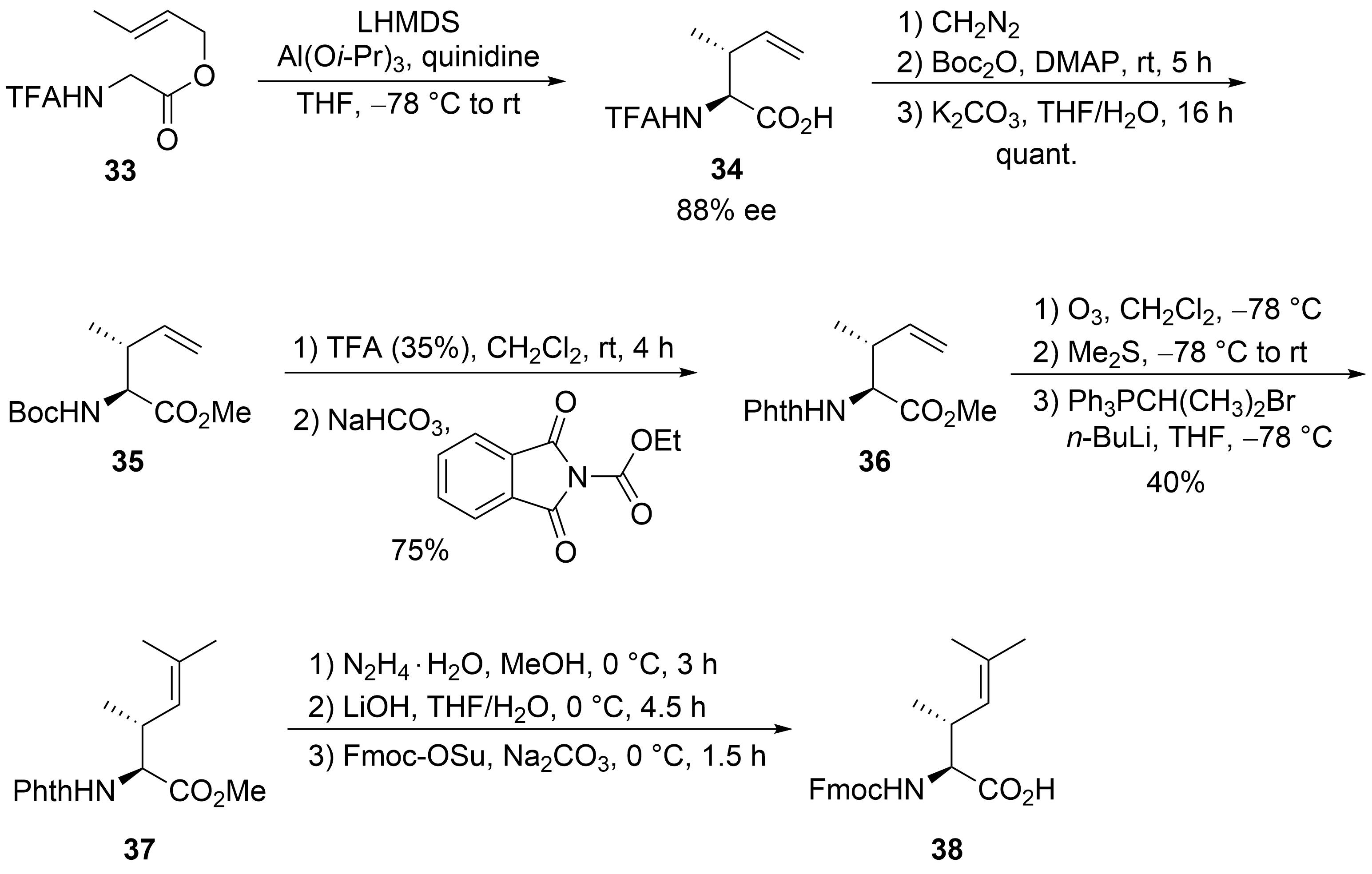
Scheme 8. Synthesis of protected dehydroamino acid 38 (building block ⑦).
After the desired building blocks were created, the synthesis of cyclomarin C and especially the best position for macrocyclization was investigated (Scheme 9)[30][39]. An attempt to align the synthesis to the biosynthetic pathway and to cyclize the linear heptapeptide precursor between the unusual tryptophan ① and the unsaturated amino acid ⑦ failed. Although obtaining the linear peptide in a [3+3+1] peptide fragment coupling strategy was straightforward, the final deprotection and ring closure yielded only trace amounts of the desired product. The same was true for attempts to cyclize the linear heptapeptide between the methoxyphenylalanine ④ and valine ⑤. The trial to cyclize between the sterically less demanding hydroxyleucine ② and alanine ③ failed early in the synthesis stage. All attempts to prolong the ①,② dipeptide at the N-terminus failed. Under the basic conditions for Fmoc-deprotection, spontaneous cyclization to the corresponding diketopiperazine occurred, comparable to the previously discussed biosynthetic side reaction, which resulted in the formation of the cyclomarazines. The ultimately successful route was the cyclization between the unsaturated amino acid ⑦ and the C-terminal N-methylleucine ⑥. The linear heptapeptide was obtained via a [4+3]-coupling strategy. An allyl ester was used as the C-terminal protecting group to avoid the basic reaction conditions required for the saponification of the C-terminal ester, which caused problems in previous cyclization attempts.
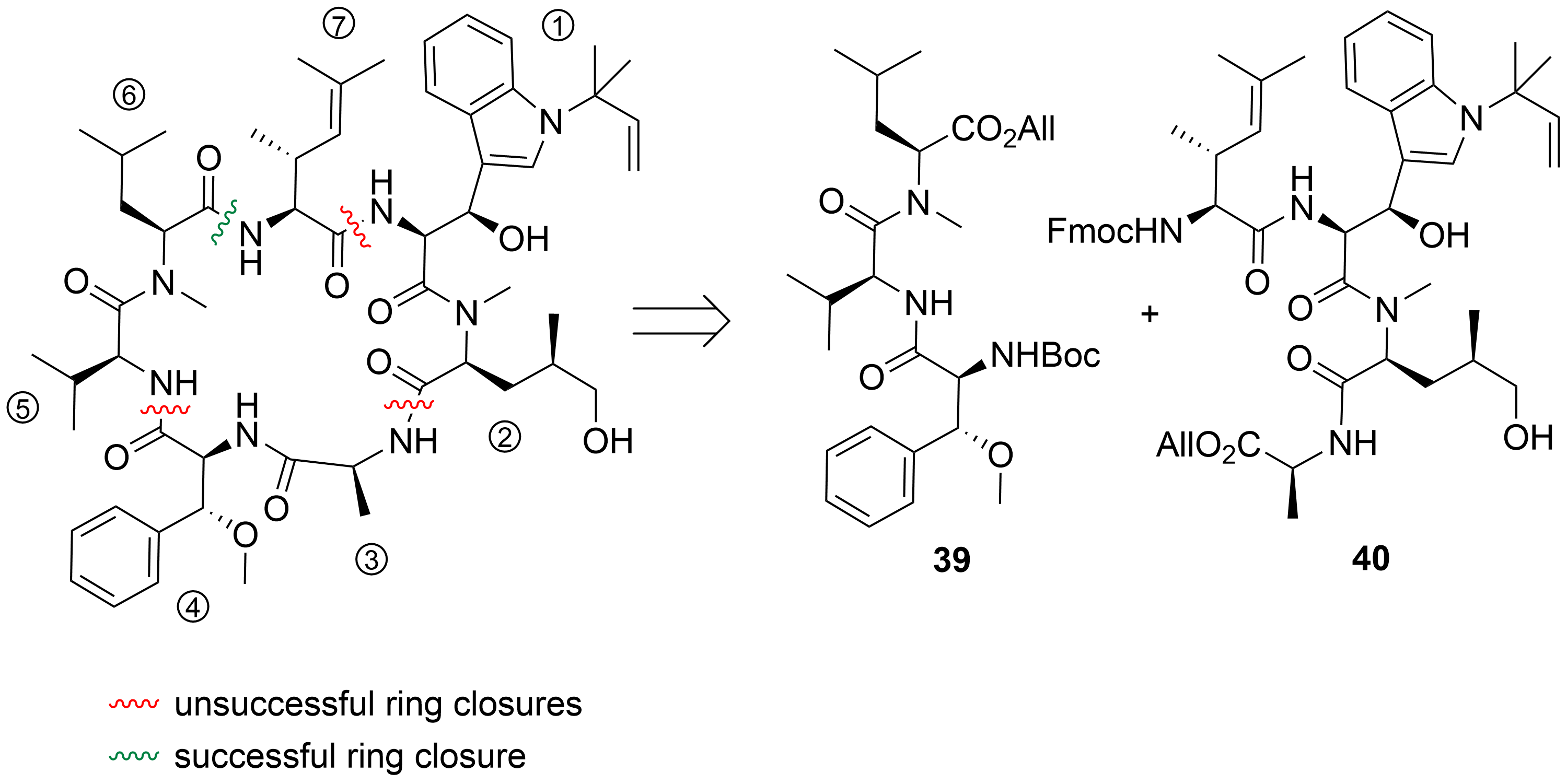
Scheme 9. Cyclization attempts for cyclomarin C[39].
The desired tri- and tetrapeptide 39 and 40 were synthesized using classical peptide coupling reactions and a combination of Boc- and Fmoc-protecting groups (Scheme 10). Because of the acid lability of β-hydroxytryptophan, Fmoc had to be used after incorporating this building block into the growing peptide chain. The synthesis of the peptide fragments was straightforward. An adequate yield of the tripeptide 39 was obtained from N-Boc-valine 41 and N-methylleucine allyl ester 42. Boc-cleavage and coupling with methoxyphenylalanine 32 produced 39, which was also N-deprotected to tripeptide 44.
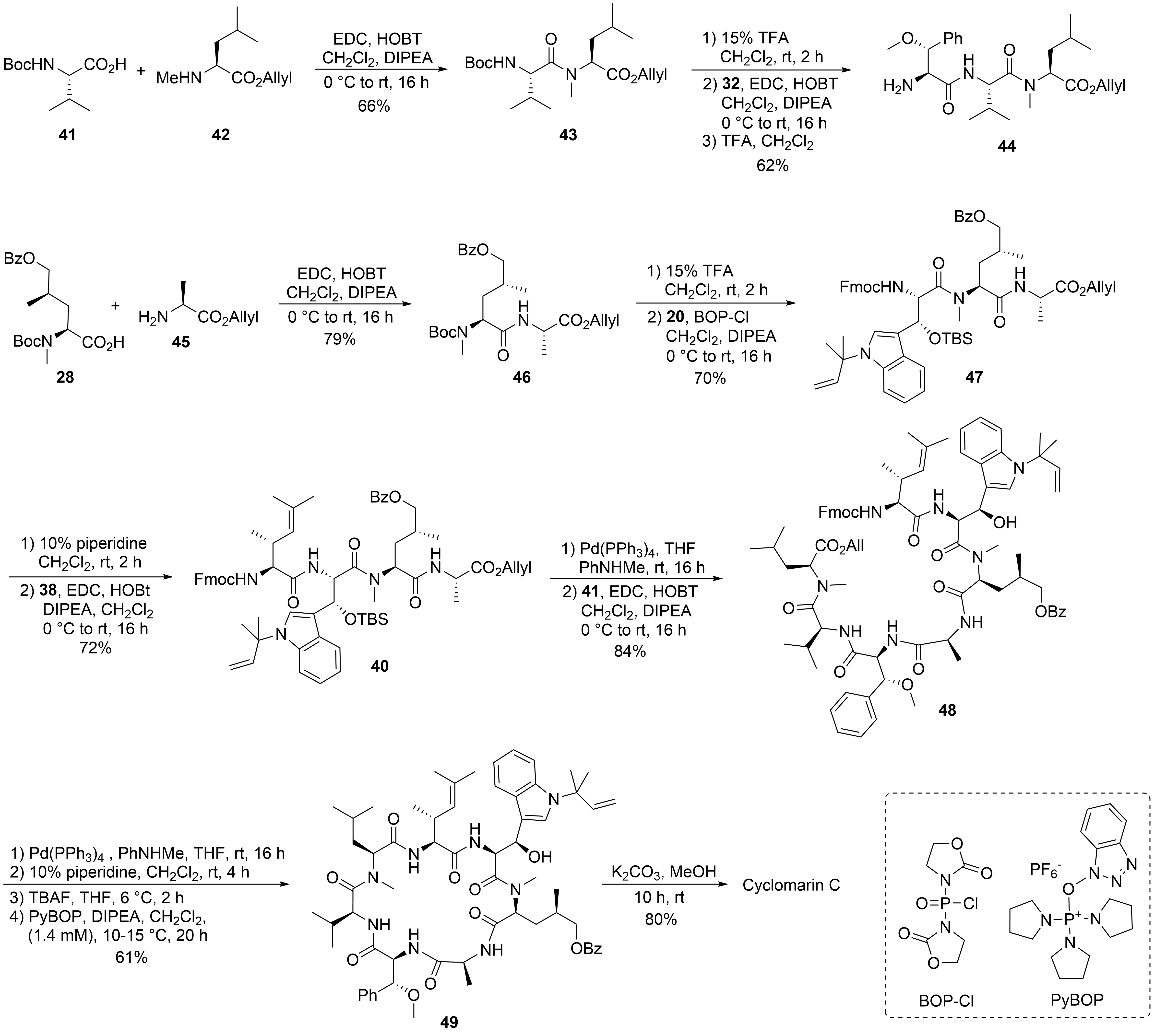
Scheme 10. Synthesis of cyclomarin C.
The synthesis of the tetrapeptide started with the coupling of protected δ-hydroxyleucine 28 with alanine allyl ester 45. After N-deprotection, the Fmoc-protected tryptophan 20 was coupled using Bop-Cl/DIPEA[40]. Careful removal of the Fmoc-protecting group from 47 and EDC/HOBT-coupling with the unsaturated building block 38 provided tetrapeptide 40. Finally, the C-terminal allyl ester was cleaved under mild Pd-catalyzed conditions, and the two peptide fragments were ready for the fragment coupling. An excellent yield of 48 was obtained using EDC/HOAt, which proved more suitable than HOBT. Subsequent deprotection of the C- and the N-terminus and removal of the OTBS-protecting group from the hydroxytryptophan provided the linear peptide precursor, which could be cyclized to 49 using PyBOP[41] under high dilution conditions and providing good yields. Finally, the benzoyl group had to be removed from the hydroxyleucine and cyclomarin C was purified via preparative HPLC.
The second synthesis of cyclomarin C and the first for cyclomarin A were reported in 2016 by Barbie and Kazmaier[42]. Both natural products differ only in the oxidation state of the prenylated β-hydroxytryptophan unit ①, which is epoxidized in cyclomarin A. Therefore, a synthetic protocol was developed which gave access to both tryptophan derivatives (Scheme 11). The synthesis started with a relatively new method for regioselective tert-prenylation of electron-demanding indoles[43]. Using indole ester 50, a palladium-catalyzed protocol delivered the required product 51 in almost quantitative yield. At 0 °C, no competitive n-prenylation was observed. In the next step, the activating ester functionality needed to be replaced by iodine. Saponification of the ester and heating the neat acid to 180 °C resulted in a clean decarboxylation to the N-prenylated indole, which could be iodinated in almost quantitative yield. Iodide 52 was used as a key building block for the synthesis of cyclomarin C, and after epoxidation, cyclomarin A. According to Yokohama et al.[44], 52 was subjected to a Sharpless dihydroxylation, which unfortunately demonstrated only moderate stereoselectivity. The best results were obtained with (DHQD)2Pyr as chiral ligand, but the ee did not exceed 80 %[45]. Subsequent tosylation of the primary OH-group and treatment with a base provided a good yield of the desired epoxide 53. The iodides 52 and 53 were next converted into organometallic reagents and reacted with a protected serinal. While the corresponding Grignard reagents provided only moderate yields and selectivities, zinc reagents were found to be superior. According to Knochel et al.[46][47], 52 was presumably converted into the indole–zinc–magnesium complex 54a, which was reacted with freshly prepared protected serinal to give the desired syn-configured 55a as a single diastereomer. In the case of the epoxyindole 53, a slightly different protocol was used. To avoid side reactions during the metalation step, 53 was lithiated at −78 °C with n-BuLi and transmetallated with ZnBr2[46][47]. The zinc reagent 54b was directly reacted with the aldehyde to create 55b. According to NMR and HPLC, only two diastereomers (ratio 9:1) could be detected following the Sharpless dihydroxylation step. Obviously, the carbonyl addition also here was highly stereoselective. Next, the secondary OH-functionality was TBS-protected under the assumption that a primary OTBS-group could be removed selectively[48]. Notably, only a combination of TBSOTf and lutidine gave the desired product 56a in high yield, while all other methods failed and resulted in the decomposition of 55a. Interestingly, no complete conversion was obtained for 55b, but the silyl ether 56b was obtained as a single stereoisomer. Obviously, the undesirable diastereomer did not undergo silylation. Next, the primary silyl protecting group was removed using NH4F in MeOH[49]. The free alcohols 57 had to be oxidized to the desired carboxylic acids 59, which were found to be highly sensitive and not very stable. By far, the best results were obtained using a two-step protocol beginning with a Parikh–Doering oxidation[50]. The also very labile aldehydes were directly oxidized to the corresponding methyl esters 58 with N-iodosuccinimide in MeOH[51]. These are stable, can be stored under standard refrigeration, and should be saponified to the free acids 59 on demand.
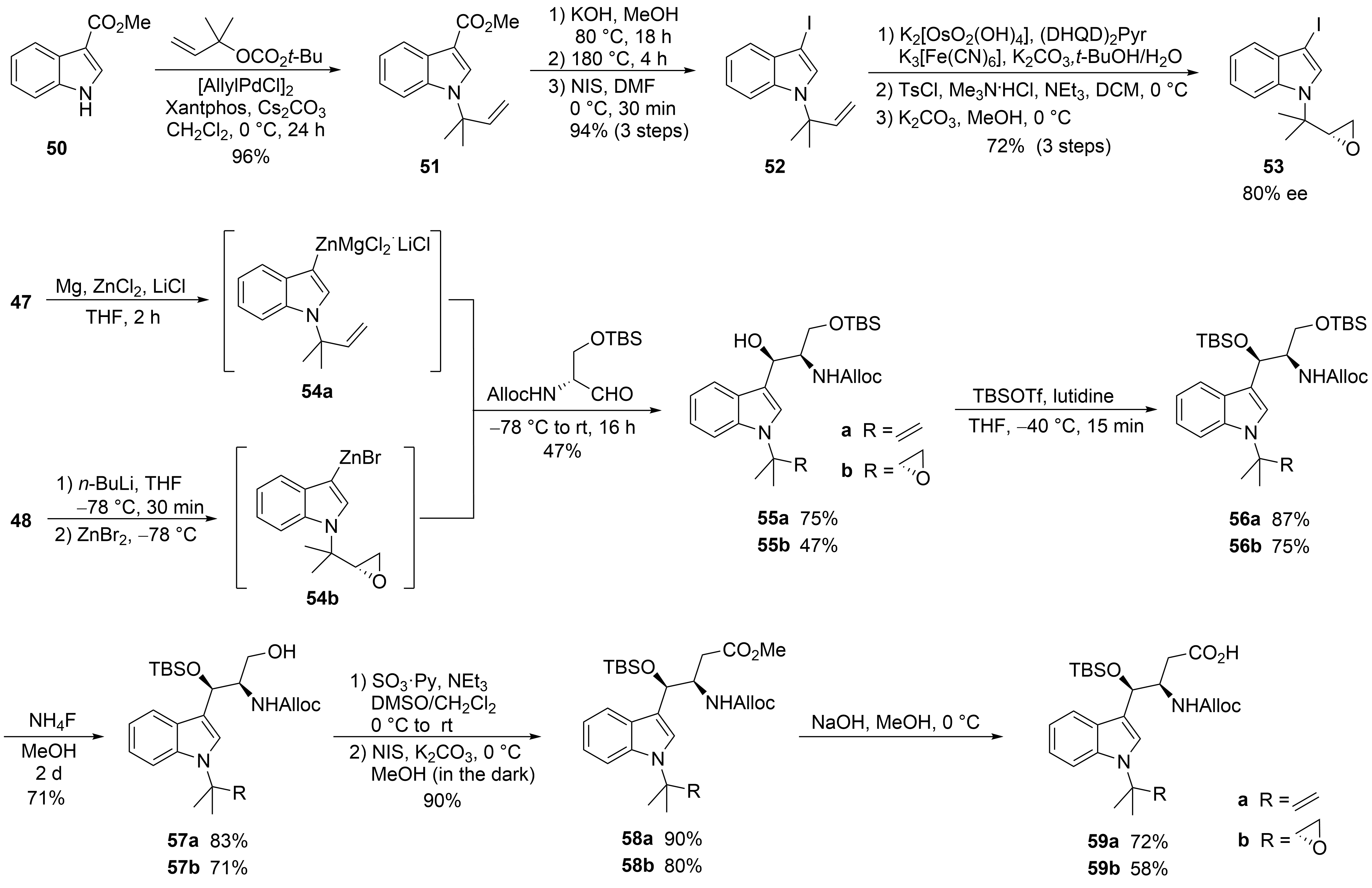
Scheme 11. Synthesis of tryptophan derivative 59 (building block ①).
A straightforward protocol was developed for the protected δ-hydroxyleucine ②, starting with the commercially available (S)-Roche ester, which was O-silylated to 60 (Scheme 12). Subsequent Dibal-H reduction provided the corresponding aldehyde, which was subjected to a Horner–Wadsworth–Emmons reaction using Schmidt’s phosphonoglycinate 61. The unsaturated amino acid 62 obtained was subjected to asymmetric hydrogenation[53] using (R)-MonoPhos as a chiral ligand[54][55]. Subsequent saponification of 63 and N-methylation yielded the desired building block 64.
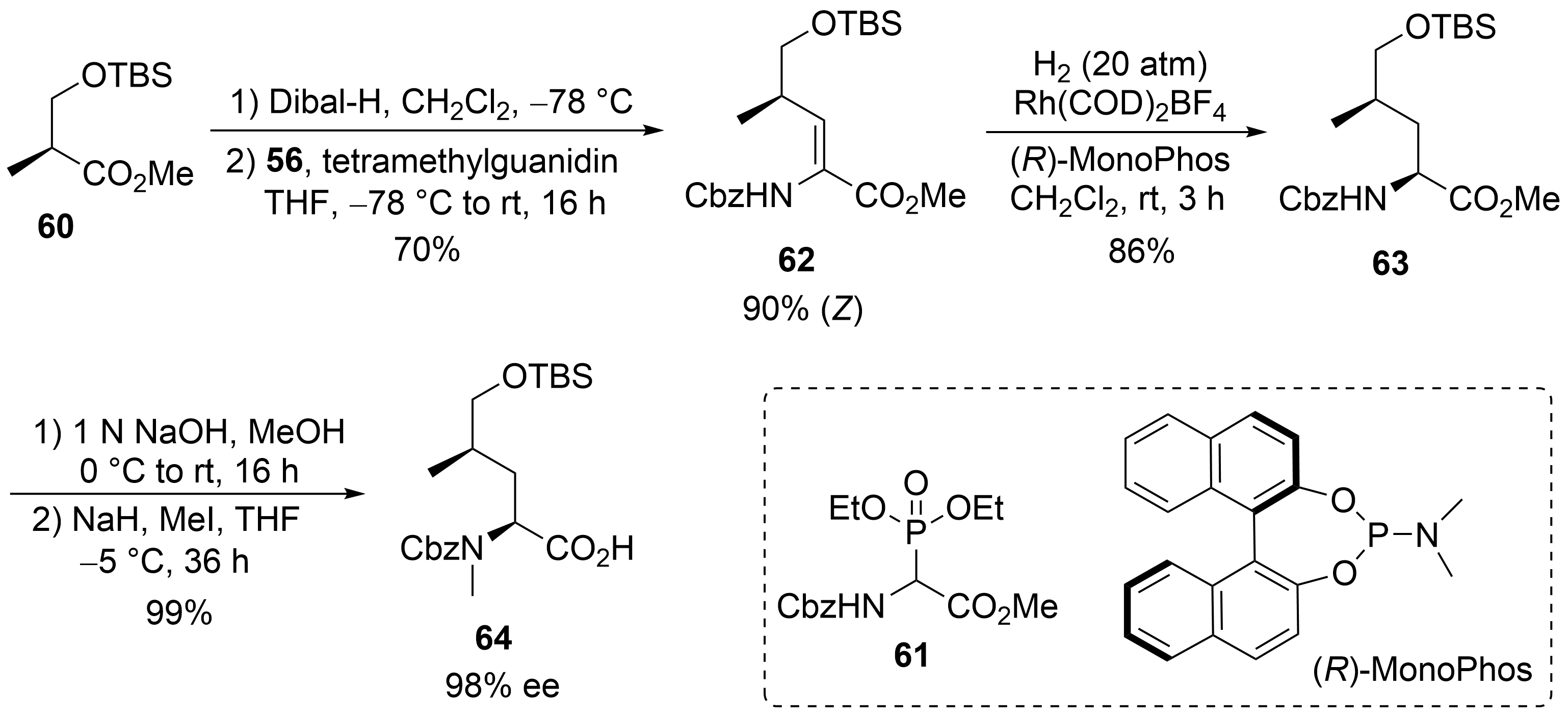
Scheme 12. Synthesis of δ-hydroxyleucine derivative 59 (building block ②).
The third unusual amino acid, β-methoxyphenylalanine ④, could be obtained similarly to the tryptophan building block ① (Scheme 13). Protected (R)-serine 65 was reduced to the corresponding aldehyde, which was subjected to a chelate-controlled aryl-metal addition. The addition of phenylmagnesium bromide provided an acceptable yield but only moderate diastereoselectivity (7:3). In contrast, in the presence of titanium salts, the desired coupling product 66 could be obtained as a single diastereomer in enantiomerically pure form. Further standard transformations yielded building block 32.
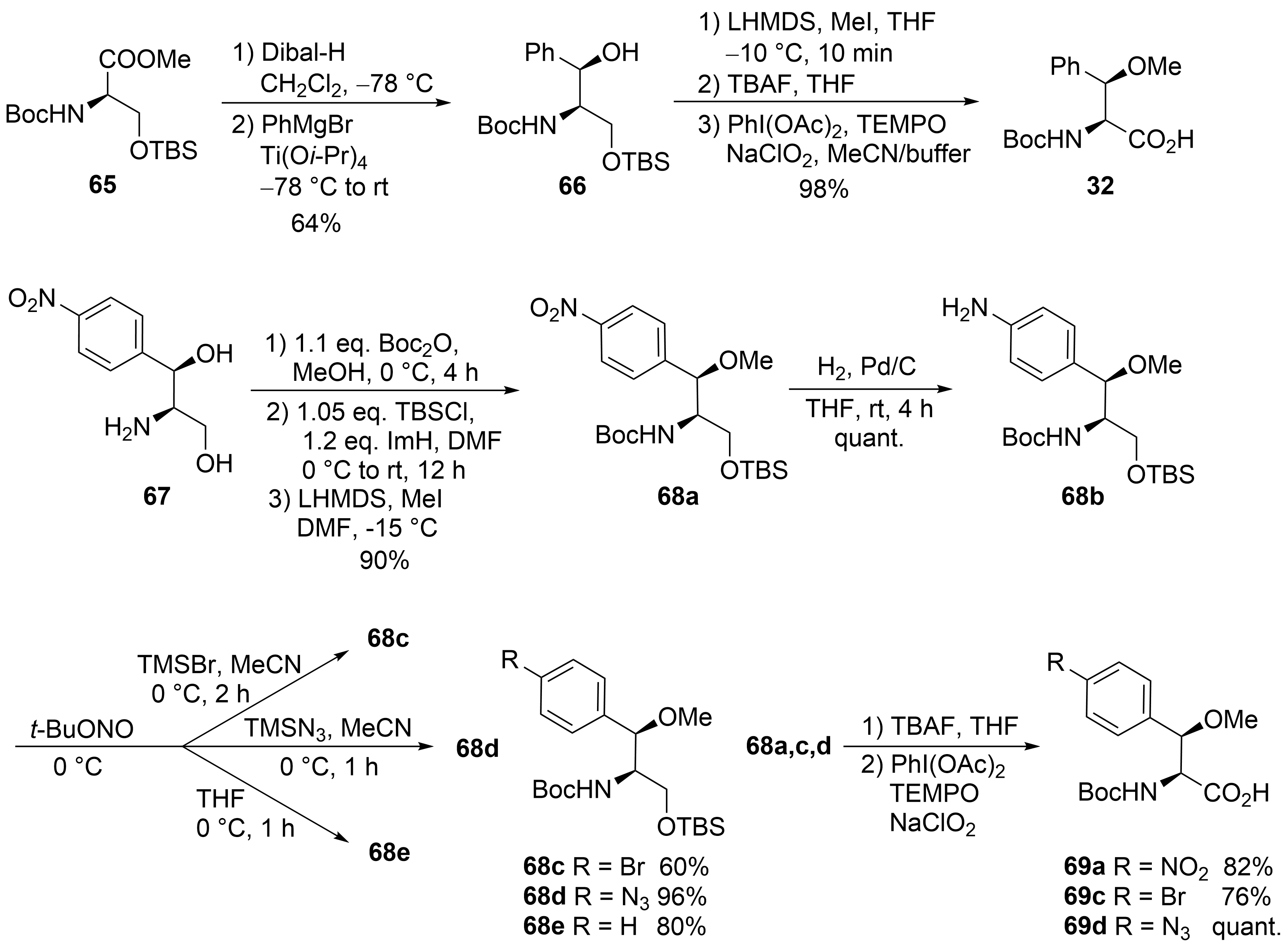
Scheme 13. Synthesis of β-methoxyphenylalanine derivatives (building block ④).
An alternative protocol, which also permitted the synthesis of substituted building blocks, was developed starting from commercially available chloramphenicol base 67 (Scheme 13)[56]. Thus, the amino diol was first converted stepwise into the protected derivative 68a. The nitro functionality could easily be reduced to the corresponding aniline derivative 68b, an ideal candidate for further structural variations via diazonium chemistry. Depending on the reaction conditions and additives used, several new derivatives 68c-e could be obtained, which could be oxidized to the corresponding amino acids 69a-d, while deamination of 68b provided the unsubstituted building block 32.
For the synthesis of the fourth, the unsaturated amino acid ⑦, also a chelate enolate Claisen rearrangement, was used starting with chiral ester 70 (Scheme 14)[57][58][59]. Rearrangement of the corresponding zinc enolate proceeded with complete chirality transfer and high stereoselectivity, and ester 71 was obtained after O-methylation. Ozonolysis provided the desired aldehyde, which was subjected to a Wittig reaction. However, only tiny amounts of the desired product could be obtained, even with a large excess of the Wittig reagents, which also caused the chiral aldehyde’s epimerization. Better results were obtained with a modified Julia–Kocienski reagent 72, normally used for (E)-selective olefination[60]. Under these conditions, the desired unsaturated building block 73 could be obtained almost epimerization-free. Saponification and change in the N-protecting group provided amino acid 74.
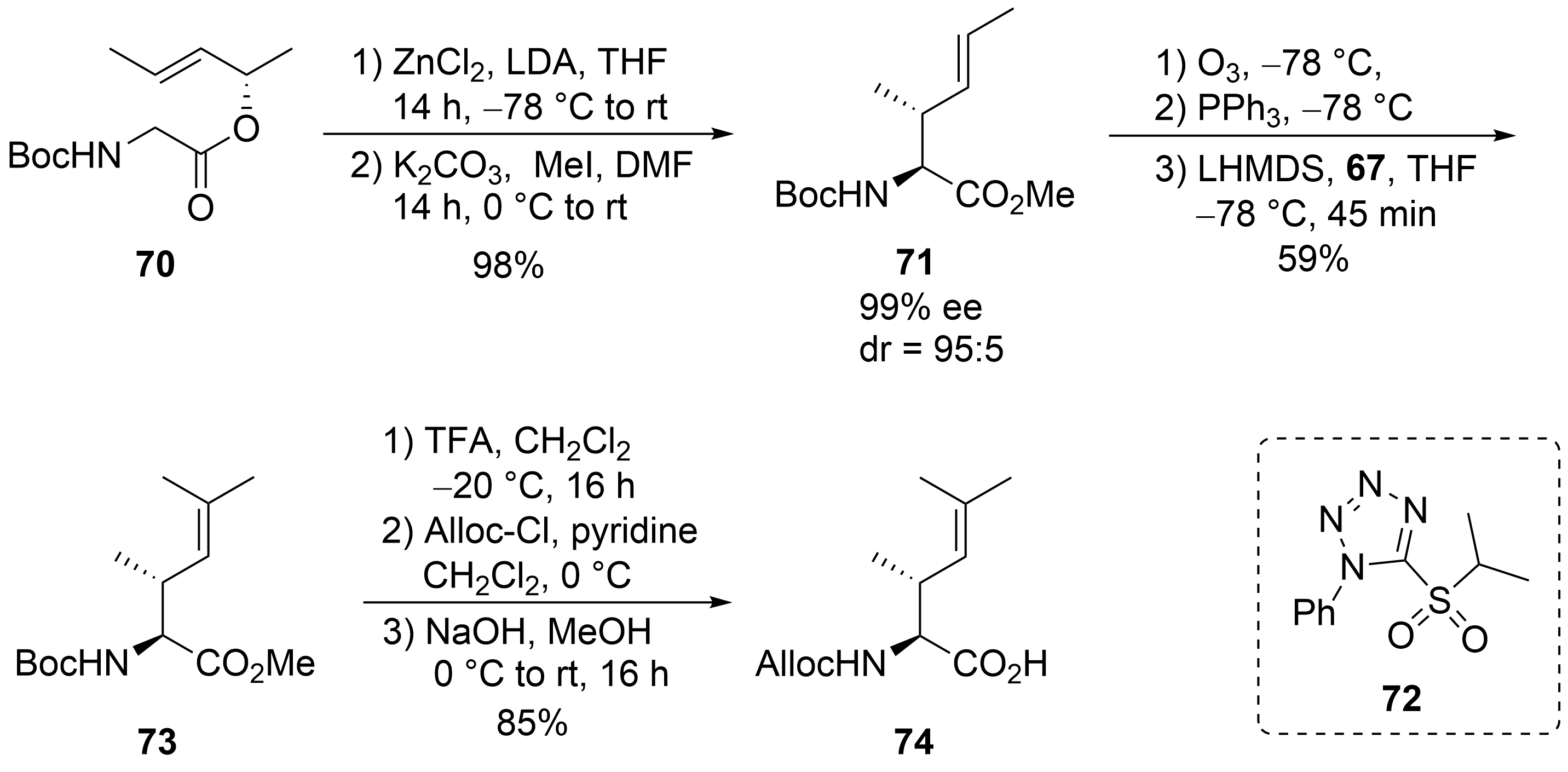
Scheme 14. Synthesis of unsaturated amino acid 69 (building block ⑦).
With all required building blocks synthesized, the linear synthesis of cyclomarins A and C started with protected N-methylleucine 75 (⑥) to cyclize the linear heptapeptide at the same position as achieved by Yao et al. (Scheme 15)[30][39]. The linear strategy was chosen to avoid epimerizations during fragment couplings, and three of the unusual building blocks were incorporated at the end of the synthesis, also allowing for modification of these building blocks and to obtain derivatives for structure-activity relationship (SAR) studies.
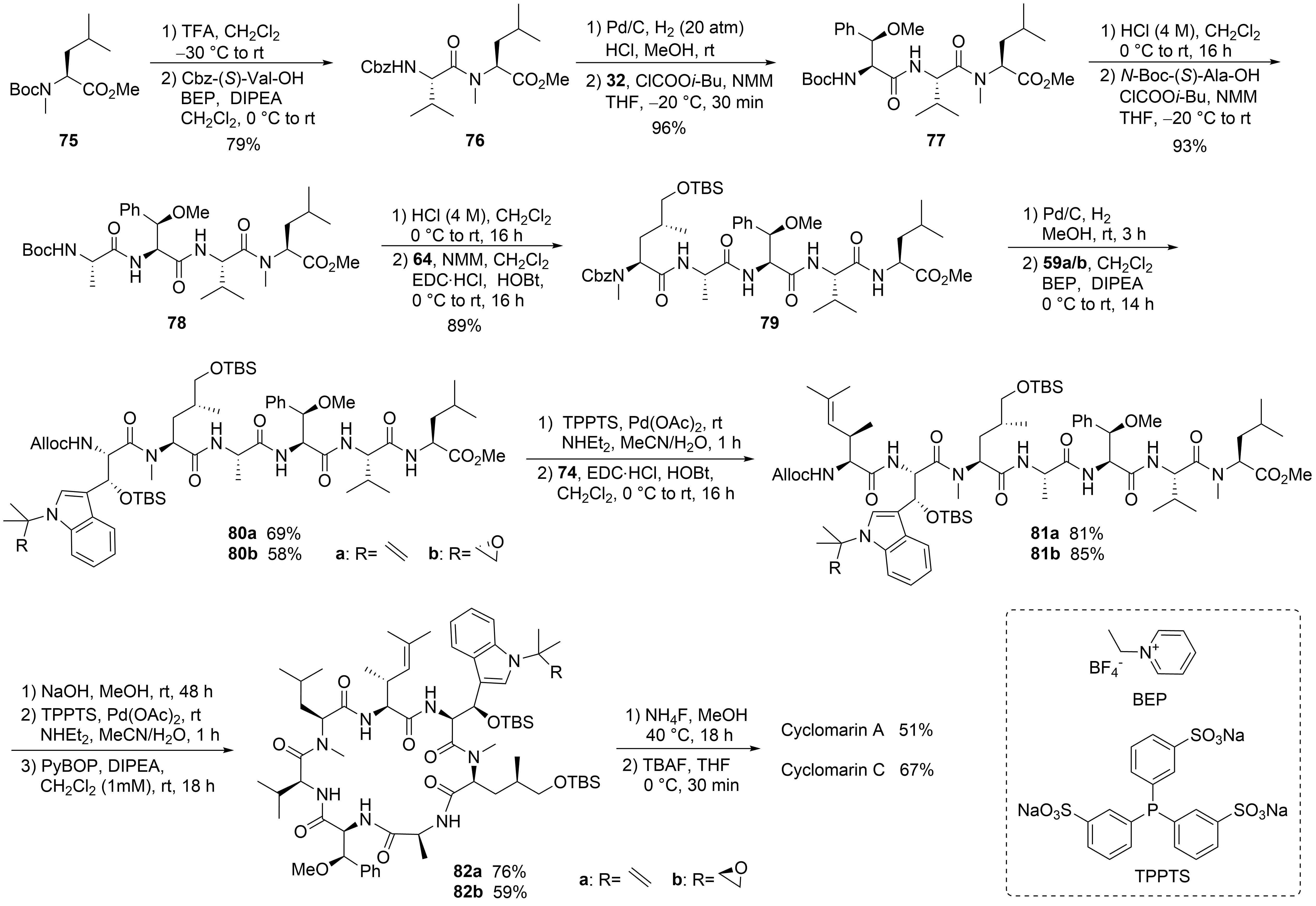
Scheme 15. Syntheses of cyclomarin A and C.
The hydrogenolysis of the Cbz-protecting group in dipeptide 76 was not a trivial task. The reaction required an H2 pressure of 20 bar to proceed, and two equivalents of HCl had to be added to avoid diketopiperazine formation. The hydrochloride salt was directly coupled with the activated amino acid 32 in the presence of excess base. The next steps were standard peptide couplings. BEP[61][62] was used to incorporate the sensitive tryptophan building blocks 59. The Alloc protecting group was removed, Pd-catalyzed, and the linear heptapeptide was cyclized using Yao’s protocol[30][39]. Finally, a two-step protocol was needed to remove the two OTBS protecting groups separately, providing good yields of cyclomarins A and C. Notably, cyclomarin D (desmethylcyclomarin C), missing only the N-methyl group of the δ-hydroxyleucin, was also obtained by this protocol[63].
References
- Nakayama, Y.; Ozawa, H.; Tahara, K.; Umezawa, H.; Studies on Ilamycin. J. Antibiot. 1962, 71, 49-50.
- Takita, T.; Ohi, K.; Maeda, K.; Okami, Y.; Umezawa, H.; New antibiotics, ilamycins. J. Antibiot. 1962, 15, 46-48.
- Higashidani, E.; Ueyanagi, J.; Shibata, M.; Nakazawa, K.; Miyake, A.; Iwasaki, H.; Yamamoto, H; Studies on Streptomycetes. 2. Rufomycin A and A, new antituberculous antibiotic. Agric. Biol. Chem. 1962, 26, 234-237.
- Shibata, M.; Yamamoto, H.; Higashidani, E.; Nakazawa, K.; Studies on Streptomycetes. 1. Streptromyces atratus nov. sp. producing new antituberculous antibiotics rufomycin A and B. Agric. Biol. Chem. 1962, 26, 228-223.
- Lewis W. Cary; Tomohisa Takita; Masako Ohnishi; A study of the secondary structure of ilamycin B1by 300 MHz proton magnetic resonance. FEBS Letters 1971, 17, 145-148, 10.1016/0014-5793(71)80584-7.
- Y. Iitaka; H. Nakamura; K. Takada; T. Takita; An X-ray study of ilamycin B1, a cyclic heptapeptide antibiotic. Acta Crystallographica Section B Structural Crystallography and Crystal Chemistry 1974, 30, 2817-2825, 10.1107/s0567740874008235.
- H. Iwasaki; B. Witkop; New Methods for Nonenzymatic Peptide Cleavage. Electrolytic, Differential, and Solvolytic Cleabage of the Antibiotic Cyclopeptide Rufomycin. Journal of the American Chemical Society 1964, 86, 4698-4708, 10.1021/ja01075a035.
- T Takita; THE AMINO ACID SEQUENCE OF ILAMYCIN AND ILAMYCIN B.. J. Antibiot. 1963, 16, 211-212.
- Takita, T.; Maeda, K.; Naganawa, H.; Umezawa, H.; Structures of ilamycin and ilamycin B2. J. Antibiot. 1964, 17, 129-131.
- Takita, T.; Naganawa, H.; Maeda, K.; Umezawa, H.; The structural diffference among ilamycin, ilamycin C1 and ilamycin C2. J. Antibiot. 1965, 18, 135-136.
- Masahiko Fujino; Takaaki Kamiya; Hidesuke Iwasaki; Jisaburo Ueyanagi; Akira Miyake; Tryptophan Moiety of Rufomycin Homologs. Chemical and Pharmaceutical Bulletin 1964, 12, 1390-1392, 10.1248/cpb.12.1390.
- T Takita; H Naganawa; K Maeda; H Umezawa; FURTHER STUDIES ON THE TRYPTOPHAN-PARTS OF ILAMYCINS.. J. Antibiot. 1964, 17, 264-265.
- Takita, T.; Naganawa, H.; L-2-Amino-4-hexenoic acid in ilamycins. J. Antibiot. 1963, 16, 246.
- Junying Ma; Hongbo Huang; Yunchang Xie; Zhiyong Liu; Jin Zhao; Chunyan Zhang; Yanxi Jia; Yun Zhang; Hua Zhang; Tianyu Zhang; et al.Jianhua Ju Biosynthesis of ilamycins featuring unusual building blocks and engineered production of enhanced anti-tuberculosis agents. Nature Communications 2017, 8, 1-10, 10.1038/s41467-017-00419-5.
- Changli Sun; Zhiyong Liu; Xiangcheng Zhu; Zhiying Fan; Xuanmei Huang; Qiaoling Wu; Xiaohong Zheng; Xiangjing Qin; Tianyu Zhang; Hua Zhang; et al.Jianhua JuJunying Ma Antitubercular Ilamycins from Marine-Derived Streptomyces atratus SCSIO ZH16 ΔilaR. Journal of Natural Products 2020, 83, 1646-1657, 10.1021/acs.jnatprod.0c00151.
- Bin Zhou; Gauri Shetye; Yang Yu; Bernard D. Santarsiero; Larry L. Klein; Cele Abad-Zapatero; Nina M. Wolf; Jinhua Cheng; Yingyu Jin; Hanki Lee; et al.Joo-Won SuhHyun LeeJonathan BissonJames B. McAlpineShao-Nong ChenSang-Hyun ChoScott G. FranzblauGuido F. Pauli Antimycobacterial Rufomycin Analogues from Streptomyces atratus Strain MJM3502. Journal of Natural Products 2020, 83, 657-667, 10.1021/acs.jnatprod.9b01095.
- Matthew K. Renner; Ya-Ching Shen; Xing-Chung Cheng; Paul R. Jensen; Walter Frankmoelle; Christopher Kauffman; William Fenical; Emil Lobkovsky; Jon Clardy; Cyclomarins A−C, New Antiinflammatory Cyclic Peptides Produced by a Marine Bacterium (Streptomycessp.). Journal of the American Chemical Society 1999, 121, 11273-11276, 10.1021/ja992482o.
- Yuzuru Mikami; Takuya Kumamoto; Hiroyuki Koshino; Daisuke Watanabe; Yuko Matsumoto; Kazuki Aoyama; Ken-Ichi Harada; Tsutomu Ishikawa; M10709, a New Cyclic Peptide Antibiotic from Clinically Isolated Streptomyces sp.. HETEROCYCLES 2010, 80, 281, 10.3987/com-09-s(s)11.
- Lei Li; Logan W. MacIntyre; Thahmina Ali; Riccardo Russo; Bimal Koirala; Yozen Hernandez; Sean F. Brady; Biosynthetic Interrogation of Soil Metagenomes Reveals Metamarin, an Uncommon Cyclomarin Congener with Activity against Mycobacterium tuberculosis. Journal of Natural Products 2021, 84, 1056-1066, 10.1021/acs.jnatprod.0c01104.
- Alexander Kiefer; Uli Kazmaier; Syntheses of Cyclomarins – Interesting Marine Natural Products with Distinct Mode of Action towards Malaria and Tuberculosis. Synthesis 2018, 51, 107-121, 10.1055/s-0037-1610377.
- Yingying Cheng; Shoubin Tang; Yian Guo; Tao Ye; Total Synthesis of Anti-tuberculosis Natural Products Ilamycins E1 and F. Organic Letters 2018, 20, 6166-6169, 10.1021/acs.orglett.8b02643.
- Michael R. Luzung; Chad Lewis; Phil S. Baran; Direct, ChemoselectiveN-tert-Prenylation of Indoles by C-H Functionalization. Angewandte Chemie International Edition 2009, 48, 7025-7029, 10.1002/anie.200902761.
- Ana Ardá; Raquel Soengas; M. Isabel Nieto; Carlos Jimenez; Jaime Rodriguez; Total Synthesis of (−)-Dysithiazolamide. Organic Letters 2008, 10, 2175-2178, 10.1021/ol800551g.
- Stephen Hanessian; Roberto Margarita; 1,3-Asymmetric induction in dianionic allylation reactions of amino acid derivatives-synthesis of functionally useful enantiopure glutamates, pipecolates and pyroglutamates. Tetrahedron Letters 1998, 39, 5887-5890, 10.1016/s0040-4039(98)00900-9.
- José M. Padrón; George Kokotos; Victor Martin; Theodoros Markidis; William A Gibbons; Vı́ctor S Martı́n; Enantiospecific synthesis of α-amino acid semialdehydes: a key step for the synthesis of unnatural unsaturated and saturated α-amino acids. Tetrahedron: Asymmetry 1998, 9, 3381-3394, 10.1016/s0957-4166(98)00354-1.
- Ulrich Schöllkopf; Ulrich Groth; Chuanzheng Deng; Enantioselective Syntheses of(R)-Amino Acids UsingL-Valine as Chiral Agent. Angewandte Chemie International Edition 1981, 20, 798-799, 10.1002/anie.198107981.
- Fernando Albericio; Marta Cases; Jordi Alsina; Salvatore A. Triolo; Louis A. Carpino; Steven A. Kates; On the use of PyAOP, a phosphonium salt derived from HOAt, in solid-phase peptide synthesis. Tetrahedron Letters 1997, 38, 4853-4856, 10.1016/s0040-4039(97)01011-3.
- Shaoqing Chen; Jiecheng Xu; Pentafluorophenyl diphenylphosphinate a new efficient coupling reagent in peptide chemistry. Tetrahedron Letters 1991, 32, 6711-6714, 10.1016/s0040-4039(00)93583-4.
- Kurapati Sathish; Gangireddy Pavan Kumar Reddy; Prathama S. Mainkar; Srivari Chandrasekhar; Synthesis of the ‘southern’ tripeptide of Cyclomarins A and C having novel anti-tuberculocidal mode of action. Tetrahedron: Asymmetry 2011, 22, 1568-1573, 10.1016/j.tetasy.2011.08.026.
- Shi-Jun Wen; Zhu-Jun Yao; Total Synthesis of Cyclomarin C. Organic Letters 2004, 6, 2721-2724, 10.1021/ol049065n.
- Shi-Jun Wen; Hong-Wang Zhang; Zhu-Jun Yao; Synthesis of a fully protected (2S,3R)-N-(1′,1′-dimethyl-2′- propenyl)-3-hydroxytryptophan from tryptophan. Tetrahedron Letters 2002, 43, 5291-5294, 10.1016/s0040-4039(02)01043-2.
- Beata Tao; Gunther Schlingloff; K.Barry Sharpless; Reversal of regioselection in the asymmetric aminohydroxylation of cinnamates. Tetrahedron Letters 1998, 39, 2507-2510, 10.1016/s0040-4039(98)00350-5.
- Philipp Barbie; Uli Kazmaier; Synthesis of fully protected, reverse N-prenylated (2S,3R)-3-hydroxytryptophan, a unique building block of the cyclomarins. Organic & Biomolecular Chemistry 2015, 13, 9267-9275, 10.1039/C5OB01438G.
- David A. Evans; David H. B. Ripin; And David P. Halstead; Kevin R. Campos; Synthesis and Absolute Stereochemical Assignment of (+)-Miyakolide. Journal of the American Chemical Society 1999, 121, 6816-6826, 10.1021/ja990789h.
- Cj Easton; Ca Hutton; Pd Roselt; Ert Tiekink; Synthesis and Molecular Structure of Stable Derivatives of (E)- and (Z)-Dehydrophenylalanine. Australian Journal of Chemistry 1991, 44, 687-694, 10.1071/ch9910687.
- Christopher J. Easton; Craig A. Hutton; Peter D. Roselt; Edward R.T. Tiekink; Stereocontrolled synthesis of β-hydroxyphenylalanine and β-hydroxytyrosine derivatives. Tetrahedron 1994, 50, 7327-7340, 10.1016/s0040-4020(01)85256-x.
- Uli Kazmaier; Achim Krebs; Synthesis of Chiralγ,δ-Unsaturated Amino Acids by Asymmetric Ester Enolate Claisen Rearrangement. Angewandte Chemie International Edition 1995, 34, 2012-2014, 10.1002/anie.199520121.
- Uli Kazmaier; Heike Mues; Achim Krebs; Asymmetric Chelated Claisen Rearrangements in the Presence of Chiral Ligands—Scope and Limitations. Chemistry – A European Journal 2002, 8, 1850-1855, 10.1002/1521-3765(20020415)8:8<1850::aid-chem1850>3.0.co;2-q.
- Shi-Jun Wen; Tai-Shan Hu; Zhu-Jun Yao; Macrocyclization studies and total synthesis of cyclomarin C, an anti-inflammatory marine cyclopeptide. Tetrahedron 2005, 61, 4931-4938, 10.1016/j.tet.2005.03.058.
- Juan Cabré; Antonio Luis Palomo; New Experimental Strategies in Amide Synthesis usingN,N-Bis[2-oxo-3-oxazolidinyl]phosphorodiamidic Chloride. Synthesis 1984, 1984, 413-417, 10.1055/s-1984-30857.
- J. Coste; D. Le-Nguyen; B. Castro; PyBOP®: A new peptide coupling reagent devoid of toxic by-product. Tetrahedron Letters 1990, 31, 205-208, 10.1016/s0040-4039(00)94371-5.
- Philipp Barbie; Uli Kazmaier; Total Synthesis of Cyclomarin A, a Marine Cycloheptapeptide with Anti-Tuberculosis and Anti-Malaria Activity. Organic Letters 2016, 18, 204-207, 10.1021/acs.orglett.5b03292.
- Kirsten F. Johnson; Ryan Van Zeeland; Levi M. Stanley; Palladium-Catalyzed Synthesis of N-tert-Prenylindoles. Organic Letters 2013, 15, 2798-2801, 10.1021/ol4011344.
- Hideyuki Sugiyama; Takayuki Shioiri; Fumiaki Yokokawa; Syntheses of four unusual amino acids, constituents of cyclomarin A. Tetrahedron Letters 2002, 43, 3489-3492, 10.1016/s0040-4039(02)00607-x.
- Aldo Spinella; Giorgio Della Sala; Irene Izzo; A Pd-Mediated Approach to the Synthesis of an Unusual β-Hydroxytryptophan Amino Acid Constituent of Cyclomarin A. Synlett 2006, 2006, 1319-1322, 10.1055/s-2006-941560.
- Albrecht Metzger; Sebastian Bernhardt; Georg Manolikakes; Paul Knochel; MgCl2-Accelerated Addition of Functionalized Organozinc Reagents to Aldehydes, Ketones, and Carbon Dioxide. Angewandte Chemie International Edition 2010, 49, 4665-4668, 10.1002/anie.201000634.
- Fabian M. Piller; Albrecht Metzger; Matthias A. Schade; Benjamin A. Haag; Andrei Gavryushin; Paul Knochel; Preparation of Polyfunctional Arylmagnesium, Arylzinc, and Benzylic Zinc Reagents by Using Magnesium in the Presence of LiCl. Chemistry – A European Journal 2009, 15, 7192-7202, 10.1002/chem.200900575.
- E. J. Corey; A. Venkateswarlu; Protection of hydroxyl groups as tert-butyldimethylsilyl derivatives. Journal of the American Chemical Society 1972, 94, 6190-6191, 10.1021/ja00772a043.
- Shuji Futagawa; Toshishige Inui; Tetsuo Shiba; Nuclear Magnetic Resonance Study of the Stereoisomeric 2-Oxazolidone and 2-Phenyl-2-oxazoline Derivatives of α-Amino-β-hydroxy Acids. Bulletin of the Chemical Society of Japan 1973, 46, 3308-3310, 10.1246/bcsj.46.3308.
- Jekishan R. Parikh; William V. E. Doering; Sulfur trioxide in the oxidation of alcohols by dimethyl sulfoxide. Journal of the American Chemical Society 1967, 89, 5505-5507, 10.1021/ja00997a067.
- Chriss McDonald; Harald Holcomb; Kenneth Kennedy; Elijah Kirkpatrick; Todd Leathers; Penny Vanemon; The N-iodosuccinimide-mediated conversion of aldehydes to methyl esters. The Journal of Organic Chemistry 1989, 54, 1213-1215, 10.1021/jo00266a046.
- Schmidt, U.; Griesser, H.; Leitenberger, V.; Lieberknecht, A.; Mangold, R.; Meyer, R.; Riedl, B.; Diastereoselective formation of (Z)-didehydroamino acid-esters. Synthesis 1992, 1992, 487-490.
- Ulrich Schmidt; Albrecht Lieberknecht; Uli Kazmaier; Helmut Griesser; Günther Jung; Jörg Metzger; Amino Acids and Peptides; 75. Synthesis of Di- and Trihydroxyamino Acids - Construction of Lipophilic Tripalmitoyldihydroxy-α-amino Acids. Synthesis 1991, 1991, 49-55, 10.1055/s-1991-26378.
- Lavinia Panella; Alicia Marco Aleixandre; Gerlof J. Kruidhof; Jort Robertus; Bernard Feringa; Johannes G. De Vries; Adriaan Minnaard; Enantioselective Rh-Catalyzed Hydrogenation ofN-Formyl Dehydroamino Esters with Monodentate Phosphoramidite Ligands. The Journal of Organic Chemistry 2006, 71, 2026-2036, 10.1021/jo052451d.
- Michel Van Den Berg; Adriaan Minnaard; Ebe P. Schudde; Jan Van Esch; André H. M. De Vries; Johannes G. De Vries; Ben L. Feringa; Highly Enantioselective Rhodium-Catalyzed Hydrogenation with Monodentate Ligands. Journal of the American Chemical Society 2000, 122, 11539-11540, 10.1021/ja002507f.
- Alexander Kiefer; Uli Kazmaier; Synthesis of modified β-methoxyphenylalanines via diazonium chemistry and their incorporation in desoxycyclomarin analogues. Organic & Biomolecular Chemistry 2019, 17, 88-102, 10.1039/c8ob02777c.
- Uli Kazmaier; Christiane Schneider; Stereoselective Synthesis of Unsaturated Polyhydroxylated Amino Acids via Ester Enolate Claisen Rearrangement. Synlett 1996, 1996, 975-977, 10.1055/s-1996-5637.
- Uli Kazmaier; Christiane Schneider; Application of the Asymmetric Chelate Enolate Claisen Rearrangement to the Synthesis of Unsaturated Polyhydroxylated Amino Acids. Synthesis 1998, 1998, 1321-1326, 10.1055/s-1998-6104.
- Uli Kazmaier; Christiane Schneider; Application of the asymmetric chelate-enolate claise rearrangement to the synthesis of 5-epi-isofagomine. Tetrahedron Letters 1998, 39, 817-818, 10.1016/s0040-4039(97)10855-3.
- Christiane Marti; Erick Carreira; Total Synthesis of (−)-Spirotryprostatin B: Synthesis and Related Studies. Journal of the American Chemical Society 2005, 127, 11505-11515, 10.1021/ja0518880.
- Peng Li; Jie-Cheng Xu; 1-Ethyl 2-Halopyridinium Salts, Highly Efficient Coupling Reagents for Hindered Peptide Synthesis both in Solution and the Solid-Phase. Tetrahedron 2000, 56, 8119-8131, 10.1016/s0040-4020(00)00657-8.
- Peng Li; Jie Cheng Xu; 2-Bromo-1-ethyl Pyridinium Tetrafluoroborate (BEP): A Powerful Coupling Reagent forN-Methylated Peptide Synthesis. Chemistry Letters 2000, 29, 204-205, 10.1246/cl.2000.204.
- Philipp Barbie; Uli Kazmaier; Total synthesis of cyclomarins A, C and D, marine cyclic peptides with interesting anti-tuberculosis and anti-malaria activities. Organic & Biomolecular Chemistry 2016, 14, 6036-6054, 10.1039/c6ob00800c.

