Ilamycins/rufomycins and cyclomarins are marine cycloheptapeptides containing unusual amino acids. Produced by Streptomyces sp., these compounds show potent activity against a range of mycobacteria, including multidrug-resistant strains of Mycobacterium tuberculosis. The cyclomarins are also very potent inhibitors of Plasmodium falciparum. Biosynthetically the cyclopeptides are obtained via a heptamodular nonribosomal peptide synthetase (NRPS) that directly incorporates some of the nonproteinogenic amino acids. A wide range of derivatives can be obtained by fermentation, while bioengineering also allows the mutasynthesis of derivatives, especially cyclomarins. Other derivatives are accessible by semisynthesis or total synthesis, reported for both natural product classes.
- ilamycins
- rufomycins
- cyclomarins
- tuberculosis
- malaria
- cyclopeptides
- biosynthesis
- total synthesis
- natural products
1. Discovery of Anti-Tubercular Cycloheptapeptides
1.1. Discovery of the Ilamycins/Rufomycins
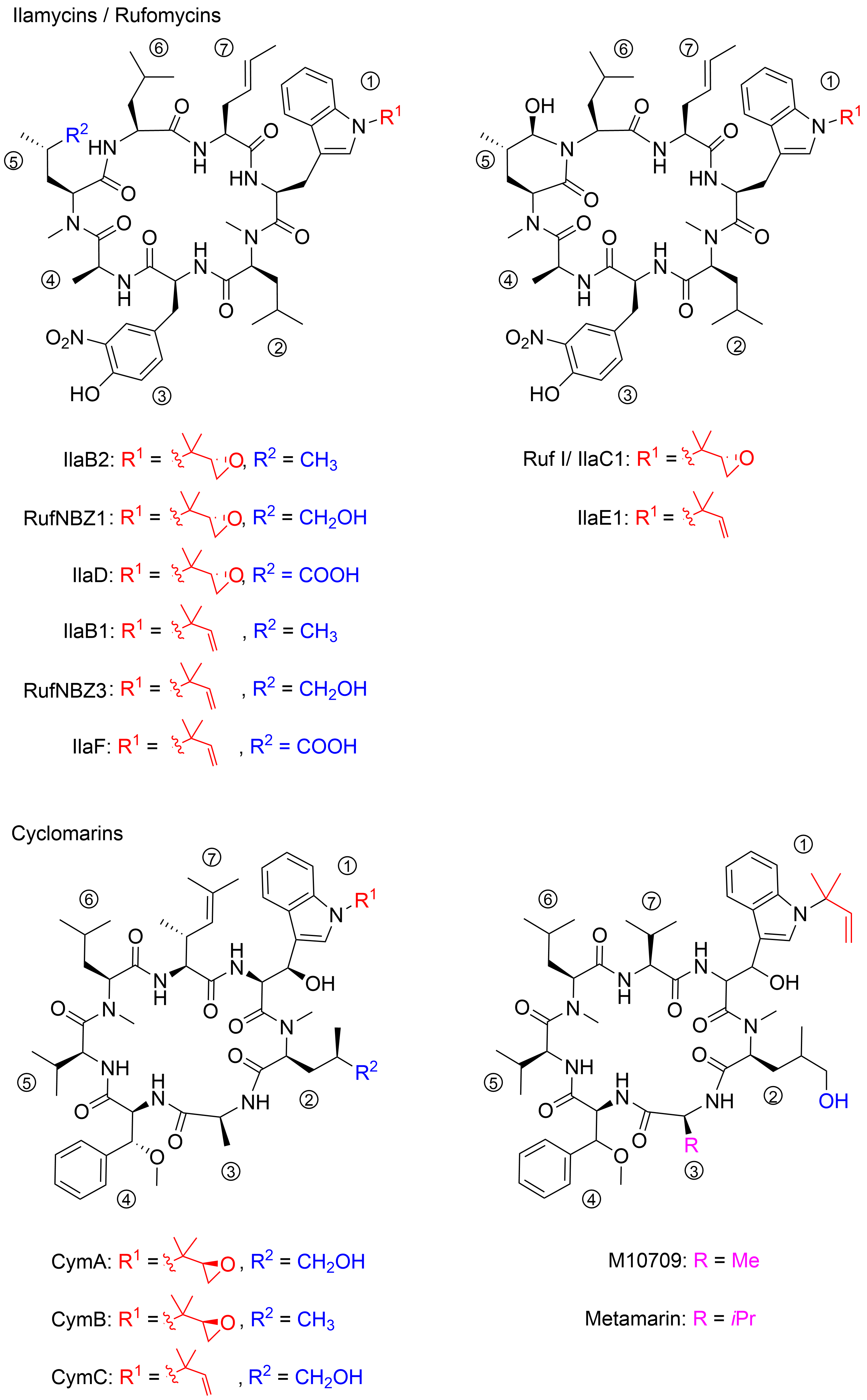
1.2. Discovery of the Cyclomarins
2. Total Syntheses of Marine Cycloheptapeptides
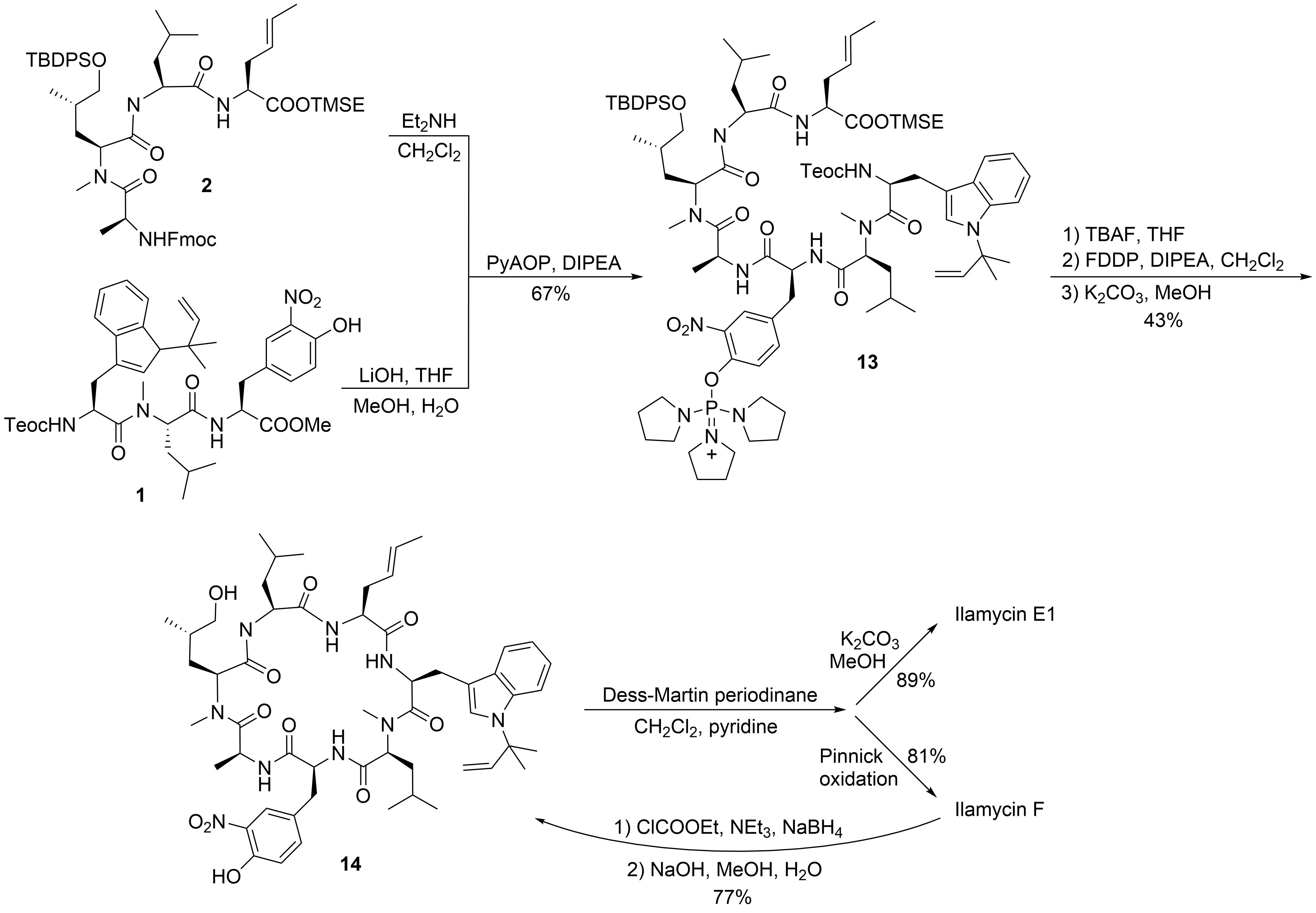
2.1. Total Synthesis of Ilamycins/Rufomycins
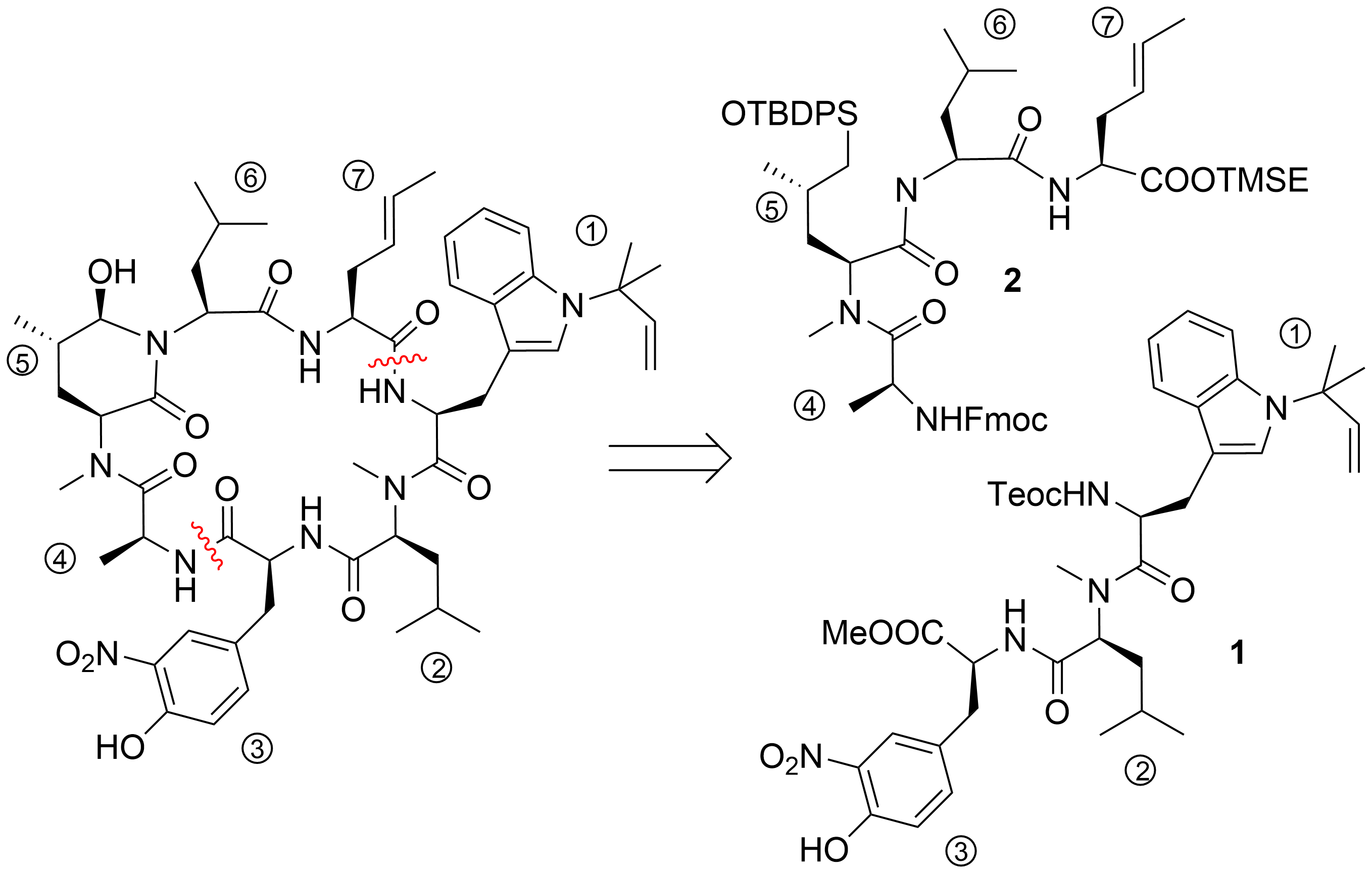
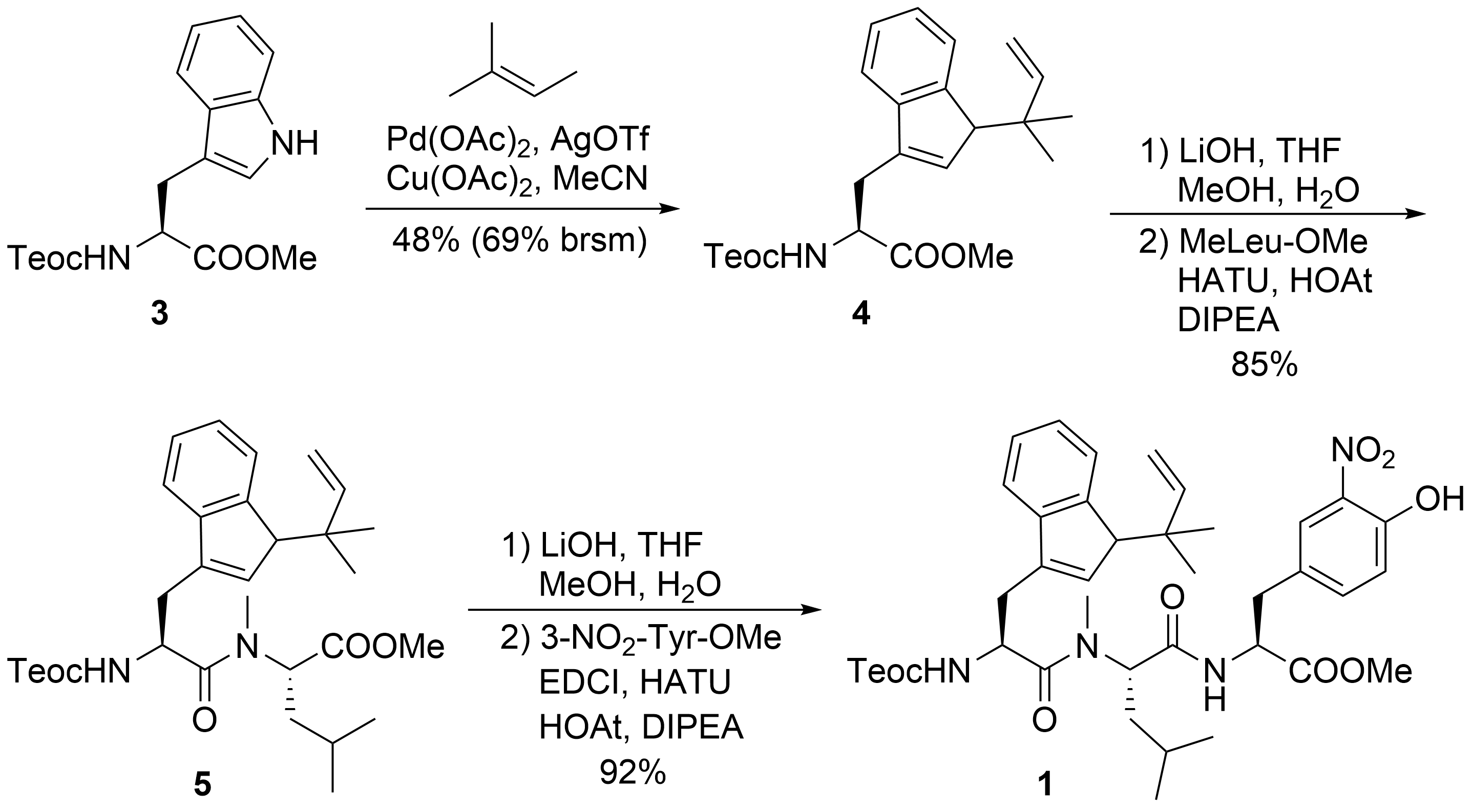
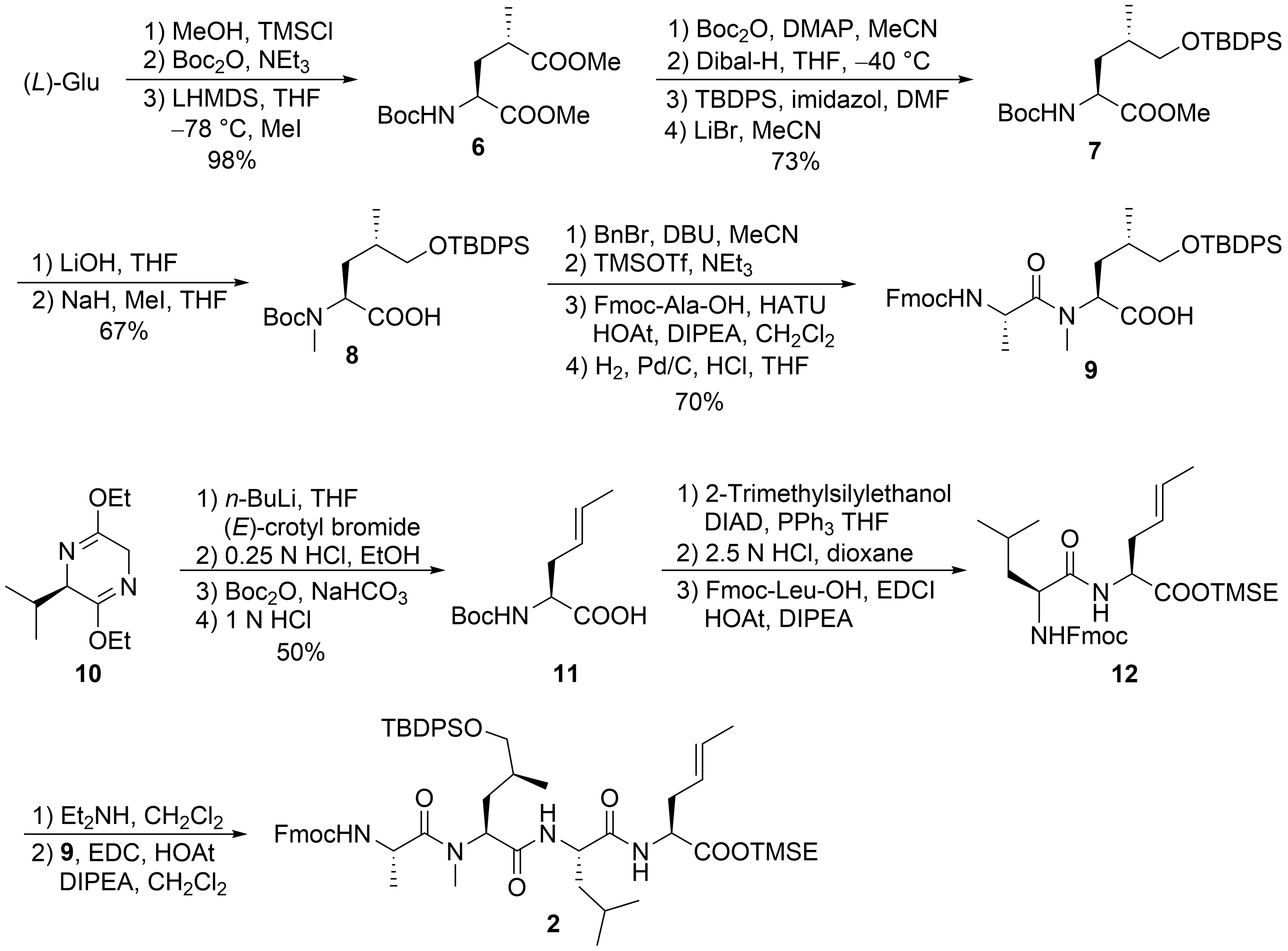
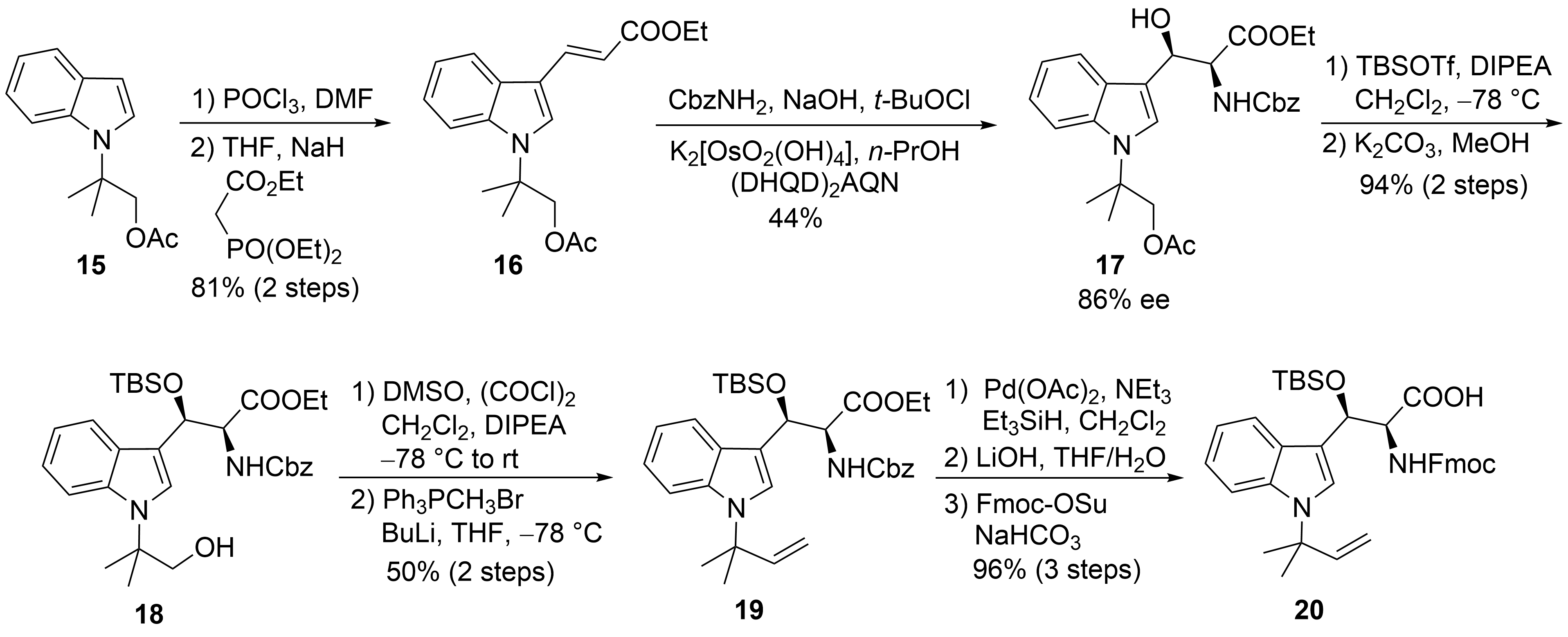
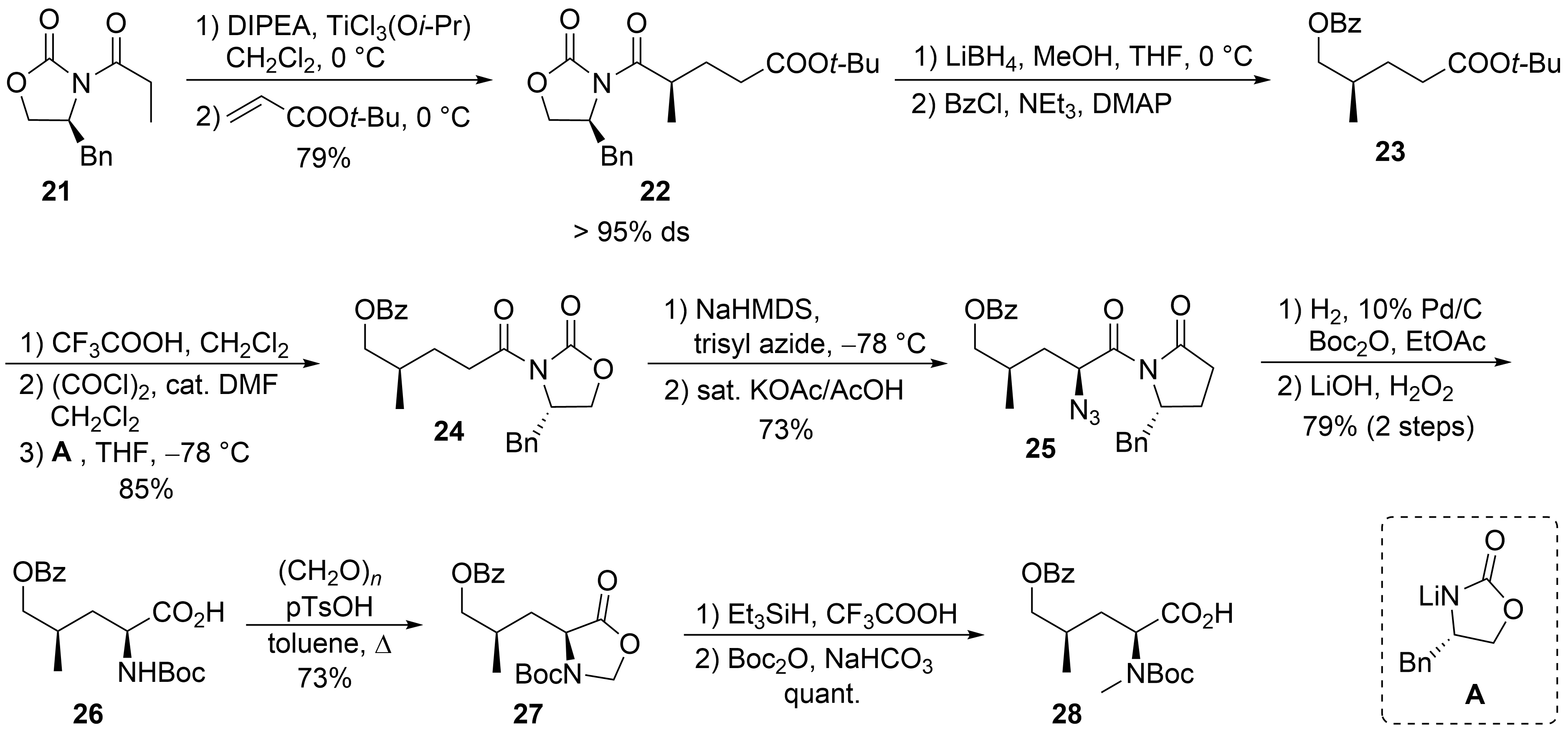
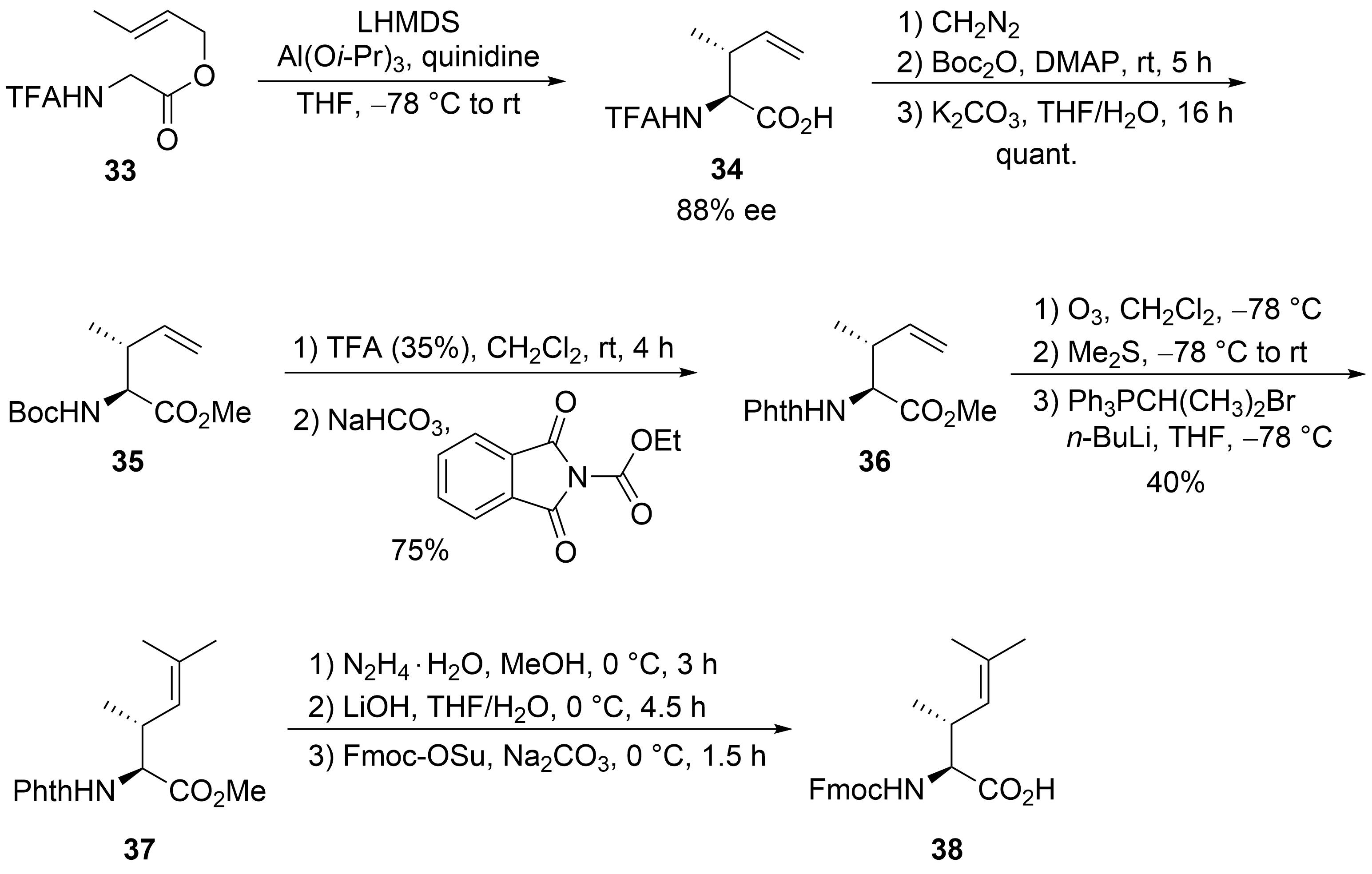
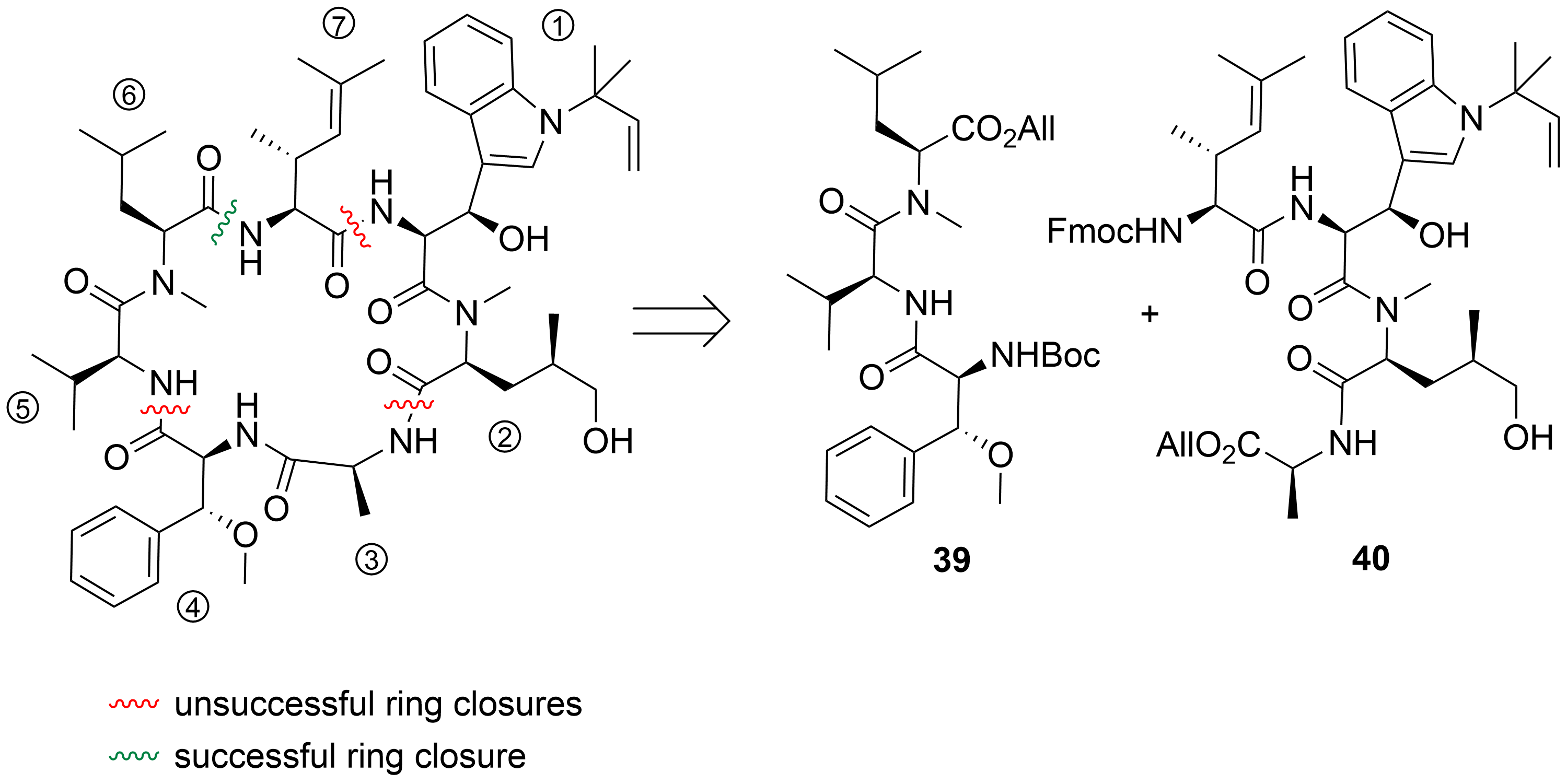
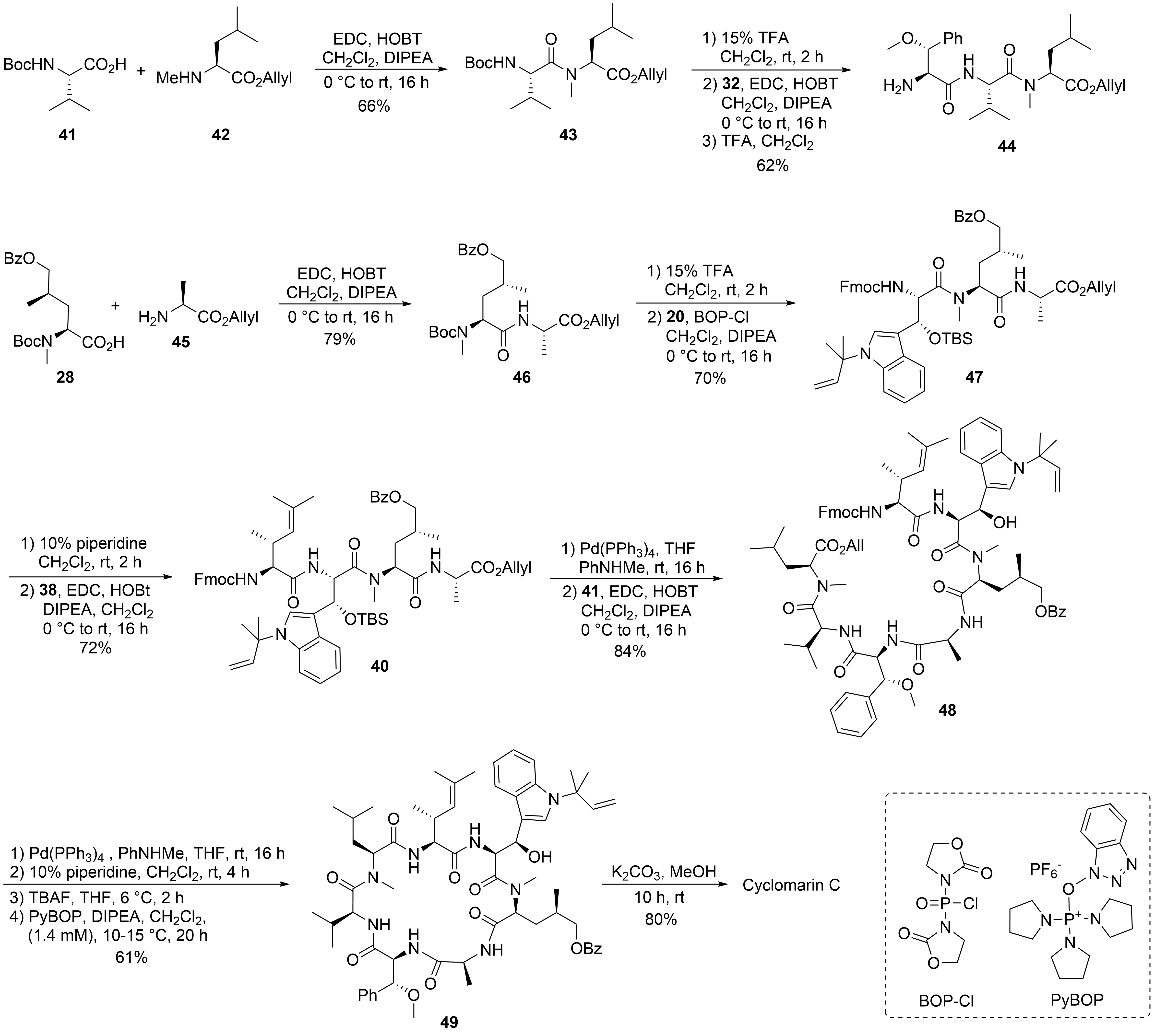
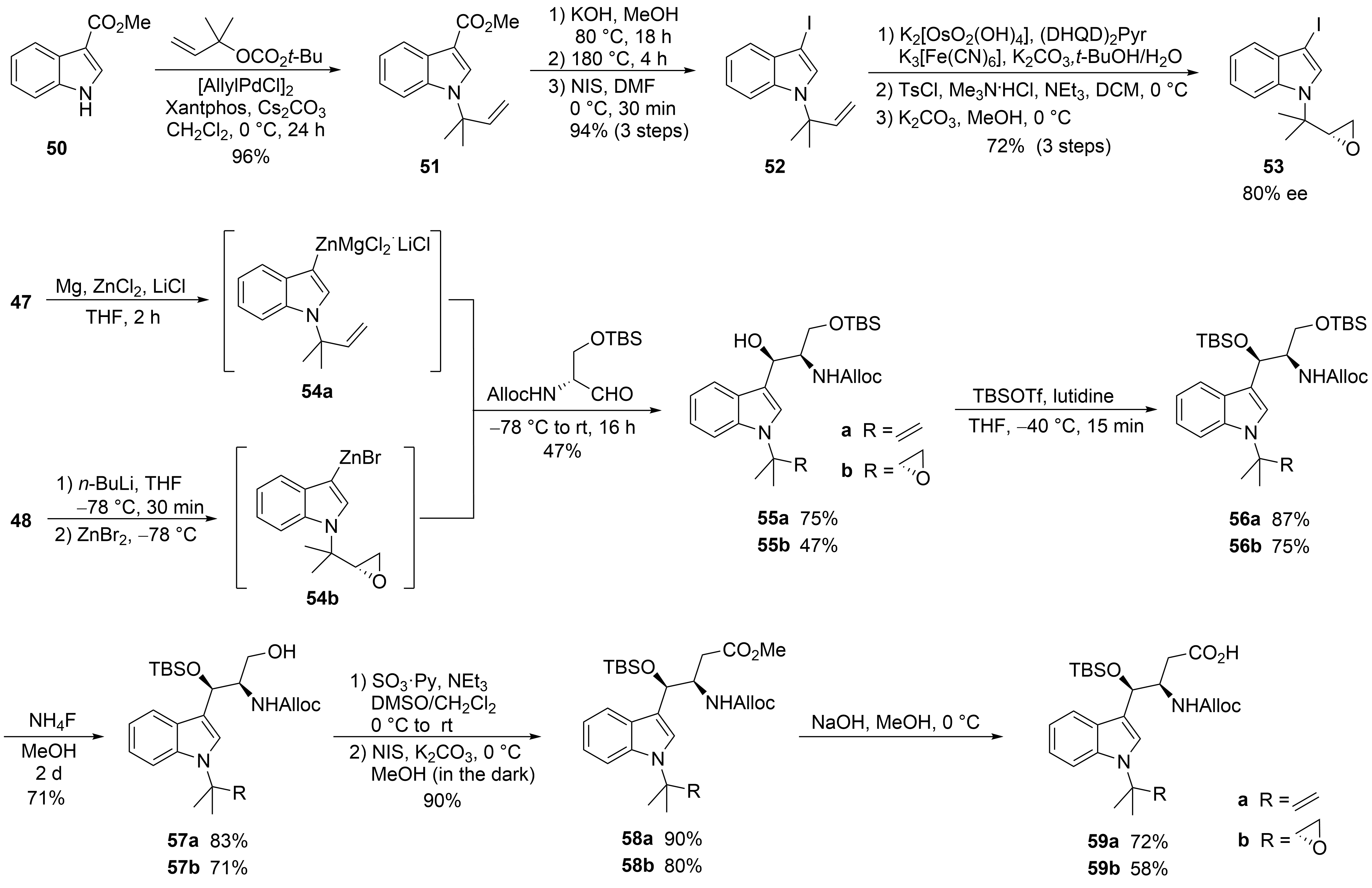
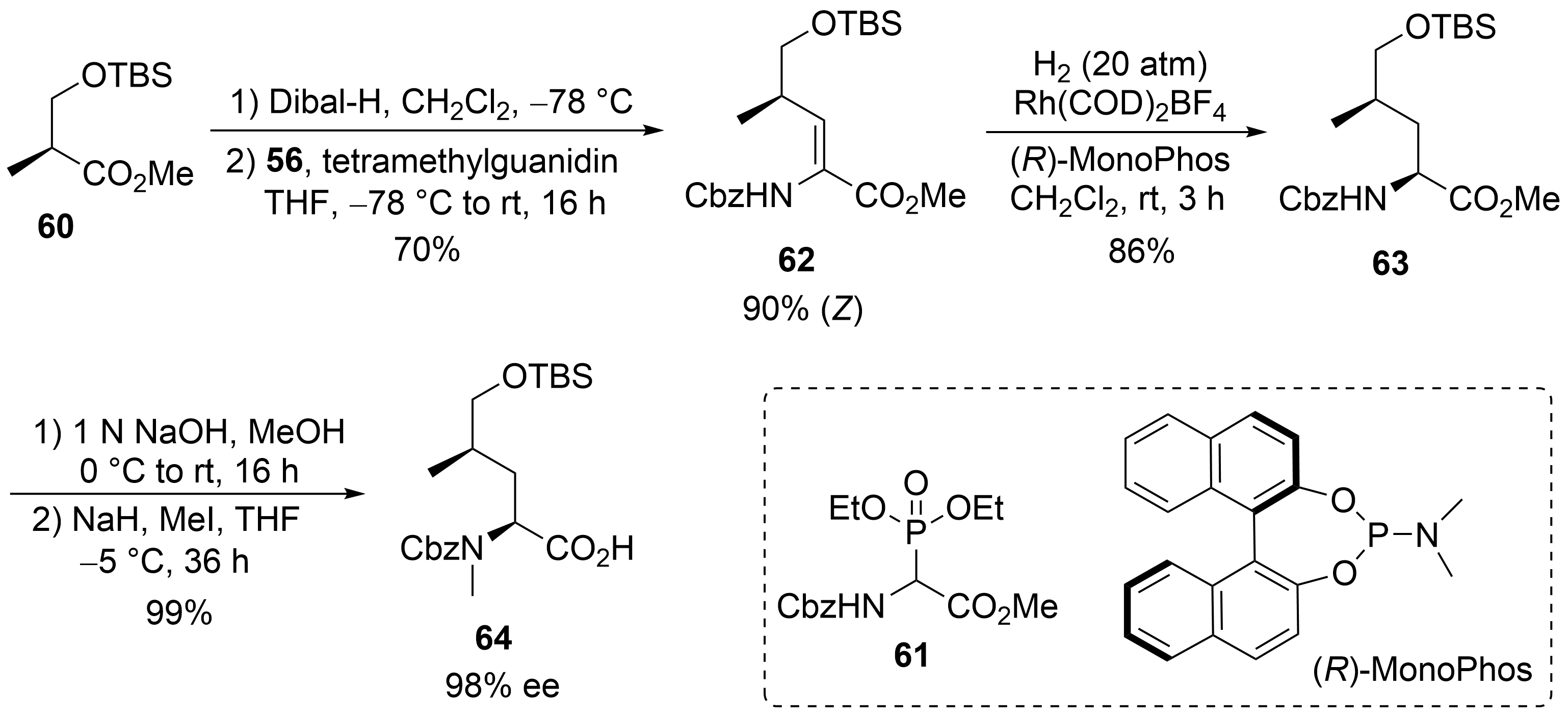
2.2. Total Synthesis of Cyclomarins

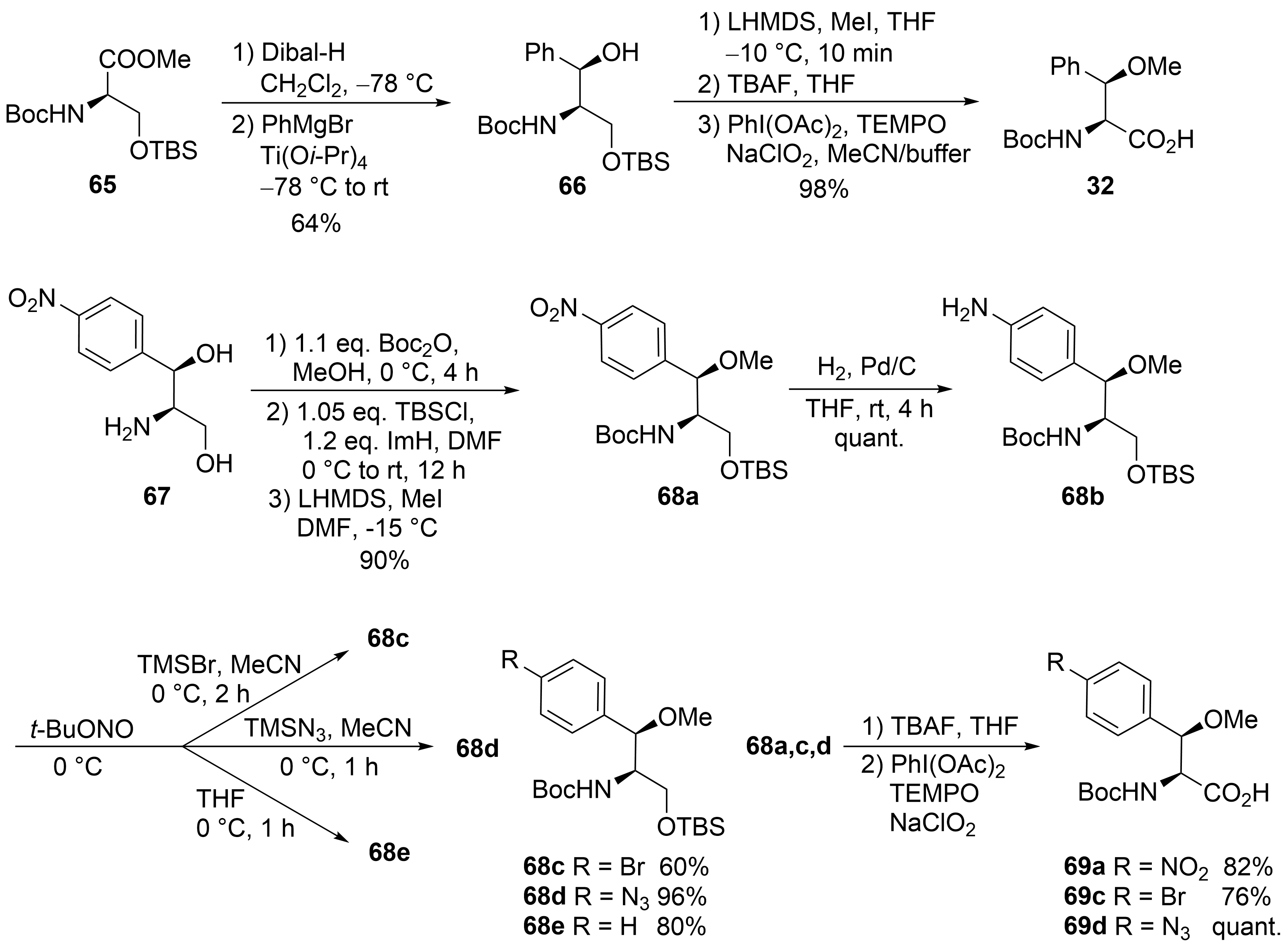
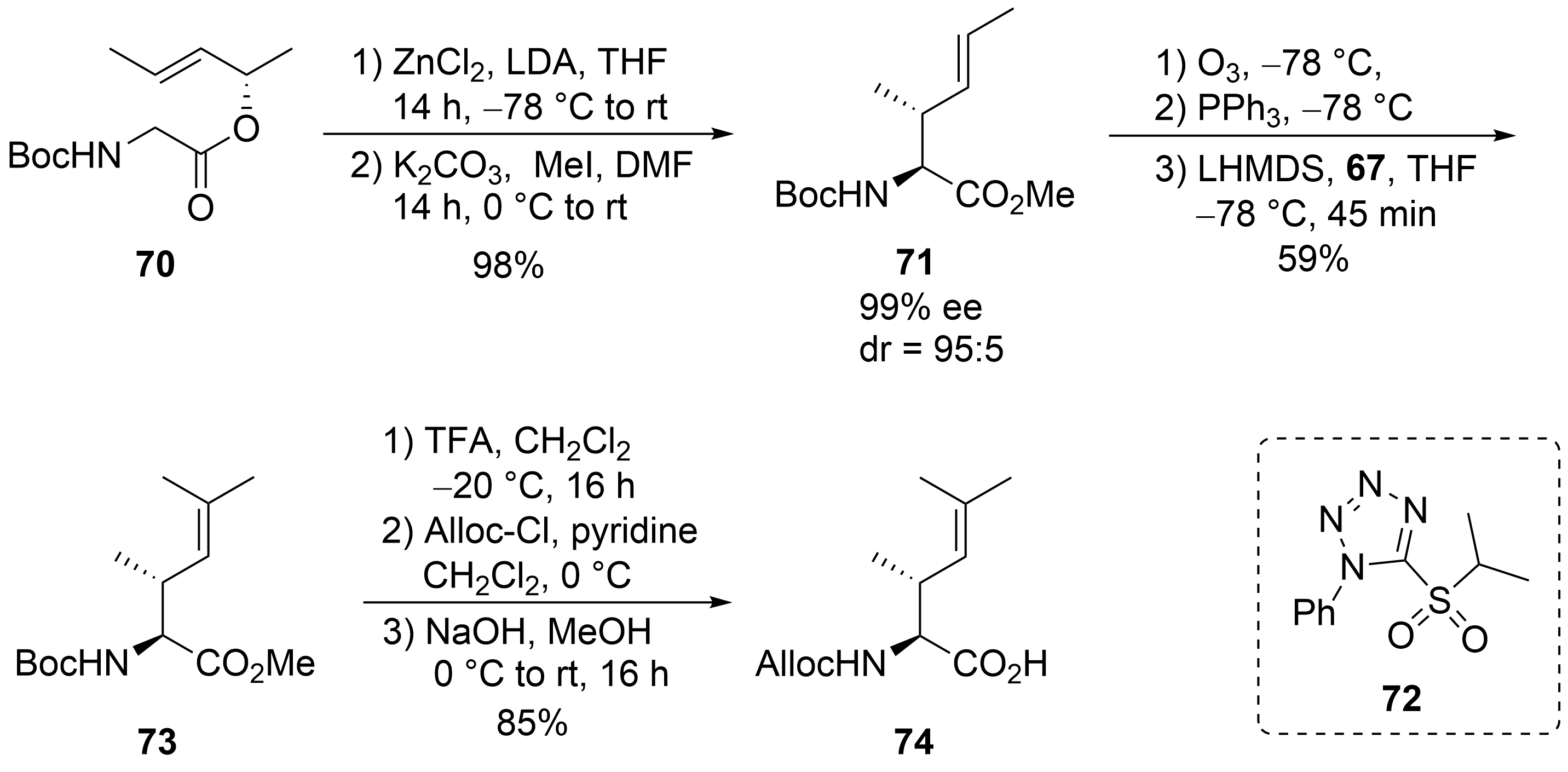
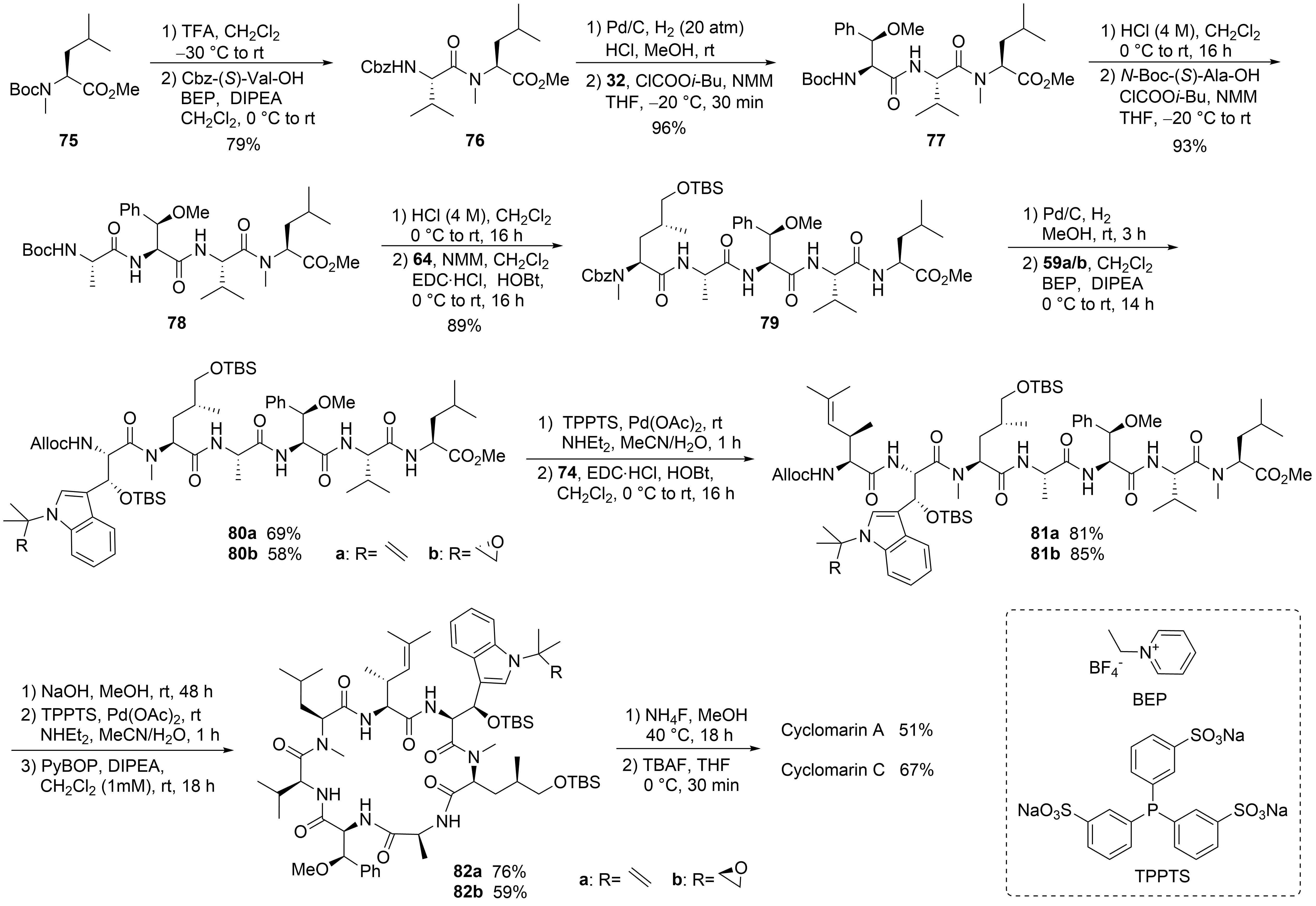
This entry is adapted from the peer-reviewed paper 10.3390/md19080446
References
- Nakayama, Y.; Ozawa, H.; Tahara, K.; Umezawa, H.; Studies on Ilamycin. J. Antibiot. 1962, 71, 49-50, .
- Takita, T.; Ohi, K.; Maeda, K.; Okami, Y.; Umezawa, H.; New antibiotics, ilamycins. J. Antibiot. 1962, 15, 46-48, .
- Higashidani, E.; Ueyanagi, J.; Shibata, M.; Nakazawa, K.; Miyake, A.; Iwasaki, H.; Yamamoto, H; Studies on Streptomycetes. 2. Rufomycin A and A, new antituberculous antibiotic. Agric. Biol. Chem. 1962, 26, 234-237, .
- Shibata, M.; Yamamoto, H.; Higashidani, E.; Nakazawa, K.; Studies on Streptomycetes. 1. Streptromyces atratus nov. sp. producing new antituberculous antibiotics rufomycin A and B. Agric. Biol. Chem. 1962, 26, 228-223, .
- Lewis W. Cary; Tomohisa Takita; Masako Ohnishi; A study of the secondary structure of ilamycin B1by 300 MHz proton magnetic resonance. FEBS Letters 1971, 17, 145-148, 10.1016/0014-5793(71)80584-7.
- Y. Iitaka; H. Nakamura; K. Takada; T. Takita; An X-ray study of ilamycin B1, a cyclic heptapeptide antibiotic. Acta Crystallographica Section B Structural Crystallography and Crystal Chemistry 1974, 30, 2817-2825, 10.1107/s0567740874008235.
- H. Iwasaki; B. Witkop; New Methods for Nonenzymatic Peptide Cleavage. Electrolytic, Differential, and Solvolytic Cleabage of the Antibiotic Cyclopeptide Rufomycin. Journal of the American Chemical Society 1964, 86, 4698-4708, 10.1021/ja01075a035.
- T Takita; THE AMINO ACID SEQUENCE OF ILAMYCIN AND ILAMYCIN B.. J. Antibiot. 1963, 16, 211-212, .
- Takita, T.; Maeda, K.; Naganawa, H.; Umezawa, H.; Structures of ilamycin and ilamycin B2. J. Antibiot. 1964, 17, 129-131, .
- Takita, T.; Naganawa, H.; Maeda, K.; Umezawa, H.; The structural diffference among ilamycin, ilamycin C1 and ilamycin C2. J. Antibiot. 1965, 18, 135-136, .
- Masahiko Fujino; Takaaki Kamiya; Hidesuke Iwasaki; Jisaburo Ueyanagi; Akira Miyake; Tryptophan Moiety of Rufomycin Homologs. Chemical and Pharmaceutical Bulletin 1964, 12, 1390-1392, 10.1248/cpb.12.1390.
- T Takita; H Naganawa; K Maeda; H Umezawa; FURTHER STUDIES ON THE TRYPTOPHAN-PARTS OF ILAMYCINS.. J. Antibiot. 1964, 17, 264-265, .
- Takita, T.; Naganawa, H.; L-2-Amino-4-hexenoic acid in ilamycins. J. Antibiot. 1963, 16, 246, .
- Junying Ma; Hongbo Huang; Yunchang Xie; Zhiyong Liu; Jin Zhao; Chunyan Zhang; Yanxi Jia; Yun Zhang; Hua Zhang; Tianyu Zhang; et al. Biosynthesis of ilamycins featuring unusual building blocks and engineered production of enhanced anti-tuberculosis agents. Nature Communications 2017, 8, 1-10, 10.1038/s41467-017-00419-5.
- Changli Sun; Zhiyong Liu; Xiangcheng Zhu; Zhiying Fan; Xuanmei Huang; Qiaoling Wu; Xiaohong Zheng; Xiangjing Qin; Tianyu Zhang; Hua Zhang; et al. Antitubercular Ilamycins from Marine-Derived Streptomyces atratus SCSIO ZH16 ΔilaR. Journal of Natural Products 2020, 83, 1646-1657, 10.1021/acs.jnatprod.0c00151.
- Bin Zhou; Gauri Shetye; Yang Yu; Bernard D. Santarsiero; Larry L. Klein; Cele Abad-Zapatero; Nina M. Wolf; Jinhua Cheng; Yingyu Jin; Hanki Lee; et al. Antimycobacterial Rufomycin Analogues from Streptomyces atratus Strain MJM3502. Journal of Natural Products 2020, 83, 657-667, 10.1021/acs.jnatprod.9b01095.
- Matthew K. Renner; Ya-Ching Shen; Xing-Chung Cheng; Paul R. Jensen; Walter Frankmoelle; Christopher Kauffman; William Fenical; Emil Lobkovsky; Jon Clardy; Cyclomarins A−C, New Antiinflammatory Cyclic Peptides Produced by a Marine Bacterium (Streptomycessp.). Journal of the American Chemical Society 1999, 121, 11273-11276, 10.1021/ja992482o.
- Yuzuru Mikami; Takuya Kumamoto; Hiroyuki Koshino; Daisuke Watanabe; Yuko Matsumoto; Kazuki Aoyama; Ken-Ichi Harada; Tsutomu Ishikawa; M10709, a New Cyclic Peptide Antibiotic from Clinically Isolated Streptomyces sp.. HETEROCYCLES 2010, 80, 281, 10.3987/com-09-s(s)11.
- Lei Li; Logan W. MacIntyre; Thahmina Ali; Riccardo Russo; Bimal Koirala; Yozen Hernandez; Sean F. Brady; Biosynthetic Interrogation of Soil Metagenomes Reveals Metamarin, an Uncommon Cyclomarin Congener with Activity against Mycobacterium tuberculosis. Journal of Natural Products 2021, 84, 1056-1066, 10.1021/acs.jnatprod.0c01104.
- Alexander Kiefer; Uli Kazmaier; Syntheses of Cyclomarins – Interesting Marine Natural Products with Distinct Mode of Action towards Malaria and Tuberculosis. Synthesis 2018, 51, 107-121, 10.1055/s-0037-1610377.
- Yingying Cheng; Shoubin Tang; Yian Guo; Tao Ye; Total Synthesis of Anti-tuberculosis Natural Products Ilamycins E1 and F. Organic Letters 2018, 20, 6166-6169, 10.1021/acs.orglett.8b02643.
- Michael R. Luzung; Chad Lewis; Phil S. Baran; Direct, ChemoselectiveN-tert-Prenylation of Indoles by C-H Functionalization. Angewandte Chemie International Edition 2009, 48, 7025-7029, 10.1002/anie.200902761.
- Ana Ardá; Raquel Soengas; M. Isabel Nieto; Carlos Jimenez; Jaime Rodriguez; Total Synthesis of (−)-Dysithiazolamide. Organic Letters 2008, 10, 2175-2178, 10.1021/ol800551g.
- Stephen Hanessian; Roberto Margarita; 1,3-Asymmetric induction in dianionic allylation reactions of amino acid derivatives-synthesis of functionally useful enantiopure glutamates, pipecolates and pyroglutamates. Tetrahedron Letters 1998, 39, 5887-5890, 10.1016/s0040-4039(98)00900-9.
- José M. Padrón; George Kokotos; Victor Martin; Theodoros Markidis; William A Gibbons; Vı́ctor S Martı́n; Enantiospecific synthesis of α-amino acid semialdehydes: a key step for the synthesis of unnatural unsaturated and saturated α-amino acids. Tetrahedron: Asymmetry 1998, 9, 3381-3394, 10.1016/s0957-4166(98)00354-1.
- Ulrich Schöllkopf; Ulrich Groth; Chuanzheng Deng; Enantioselective Syntheses of(R)-Amino Acids UsingL-Valine as Chiral Agent. Angewandte Chemie International Edition 1981, 20, 798-799, 10.1002/anie.198107981.
- Fernando Albericio; Marta Cases; Jordi Alsina; Salvatore A. Triolo; Louis A. Carpino; Steven A. Kates; On the use of PyAOP, a phosphonium salt derived from HOAt, in solid-phase peptide synthesis. Tetrahedron Letters 1997, 38, 4853-4856, 10.1016/s0040-4039(97)01011-3.
- Shaoqing Chen; Jiecheng Xu; Pentafluorophenyl diphenylphosphinate a new efficient coupling reagent in peptide chemistry. Tetrahedron Letters 1991, 32, 6711-6714, 10.1016/s0040-4039(00)93583-4.
- Kurapati Sathish; Gangireddy Pavan Kumar Reddy; Prathama S. Mainkar; Srivari Chandrasekhar; Synthesis of the ‘southern’ tripeptide of Cyclomarins A and C having novel anti-tuberculocidal mode of action. Tetrahedron: Asymmetry 2011, 22, 1568-1573, 10.1016/j.tetasy.2011.08.026.
- Shi-Jun Wen; Zhu-Jun Yao; Total Synthesis of Cyclomarin C. Organic Letters 2004, 6, 2721-2724, 10.1021/ol049065n.
- Shi-Jun Wen; Hong-Wang Zhang; Zhu-Jun Yao; Synthesis of a fully protected (2S,3R)-N-(1′,1′-dimethyl-2′- propenyl)-3-hydroxytryptophan from tryptophan. Tetrahedron Letters 2002, 43, 5291-5294, 10.1016/s0040-4039(02)01043-2.
- Beata Tao; Gunther Schlingloff; K.Barry Sharpless; Reversal of regioselection in the asymmetric aminohydroxylation of cinnamates. Tetrahedron Letters 1998, 39, 2507-2510, 10.1016/s0040-4039(98)00350-5.
- Philipp Barbie; Uli Kazmaier; Synthesis of fully protected, reverse N-prenylated (2S,3R)-3-hydroxytryptophan, a unique building block of the cyclomarins. Organic & Biomolecular Chemistry 2015, 13, 9267-9275, 10.1039/C5OB01438G.
- David A. Evans; David H. B. Ripin; And David P. Halstead; Kevin R. Campos; Synthesis and Absolute Stereochemical Assignment of (+)-Miyakolide. Journal of the American Chemical Society 1999, 121, 6816-6826, 10.1021/ja990789h.
- Cj Easton; Ca Hutton; Pd Roselt; Ert Tiekink; Synthesis and Molecular Structure of Stable Derivatives of (E)- and (Z)-Dehydrophenylalanine. Australian Journal of Chemistry 1991, 44, 687-694, 10.1071/ch9910687.
- Christopher J. Easton; Craig A. Hutton; Peter D. Roselt; Edward R.T. Tiekink; Stereocontrolled synthesis of β-hydroxyphenylalanine and β-hydroxytyrosine derivatives. Tetrahedron 1994, 50, 7327-7340, 10.1016/s0040-4020(01)85256-x.
- Uli Kazmaier; Achim Krebs; Synthesis of Chiralγ,δ-Unsaturated Amino Acids by Asymmetric Ester Enolate Claisen Rearrangement. Angewandte Chemie International Edition 1995, 34, 2012-2014, 10.1002/anie.199520121.
- Uli Kazmaier; Heike Mues; Achim Krebs; Asymmetric Chelated Claisen Rearrangements in the Presence of Chiral Ligands—Scope and Limitations. Chemistry – A European Journal 2002, 8, 1850-1855, 10.1002/1521-3765(20020415)8:8<1850::aid-chem1850>3.0.co;2-q.
- Shi-Jun Wen; Tai-Shan Hu; Zhu-Jun Yao; Macrocyclization studies and total synthesis of cyclomarin C, an anti-inflammatory marine cyclopeptide. Tetrahedron 2005, 61, 4931-4938, 10.1016/j.tet.2005.03.058.
- Juan Cabré; Antonio Luis Palomo; New Experimental Strategies in Amide Synthesis usingN,N-Bis[2-oxo-3-oxazolidinyl]phosphorodiamidic Chloride. Synthesis 1984, 1984, 413-417, 10.1055/s-1984-30857.
- J. Coste; D. Le-Nguyen; B. Castro; PyBOP®: A new peptide coupling reagent devoid of toxic by-product. Tetrahedron Letters 1990, 31, 205-208, 10.1016/s0040-4039(00)94371-5.
- Philipp Barbie; Uli Kazmaier; Total Synthesis of Cyclomarin A, a Marine Cycloheptapeptide with Anti-Tuberculosis and Anti-Malaria Activity. Organic Letters 2016, 18, 204-207, 10.1021/acs.orglett.5b03292.
- Kirsten F. Johnson; Ryan Van Zeeland; Levi M. Stanley; Palladium-Catalyzed Synthesis of N-tert-Prenylindoles. Organic Letters 2013, 15, 2798-2801, 10.1021/ol4011344.
- Hideyuki Sugiyama; Takayuki Shioiri; Fumiaki Yokokawa; Syntheses of four unusual amino acids, constituents of cyclomarin A. Tetrahedron Letters 2002, 43, 3489-3492, 10.1016/s0040-4039(02)00607-x.
- Aldo Spinella; Giorgio Della Sala; Irene Izzo; A Pd-Mediated Approach to the Synthesis of an Unusual β-Hydroxytryptophan Amino Acid Constituent of Cyclomarin A. Synlett 2006, 2006, 1319-1322, 10.1055/s-2006-941560.
- Albrecht Metzger; Sebastian Bernhardt; Georg Manolikakes; Paul Knochel; MgCl2-Accelerated Addition of Functionalized Organozinc Reagents to Aldehydes, Ketones, and Carbon Dioxide. Angewandte Chemie International Edition 2010, 49, 4665-4668, 10.1002/anie.201000634.
- Fabian M. Piller; Albrecht Metzger; Matthias A. Schade; Benjamin A. Haag; Andrei Gavryushin; Paul Knochel; Preparation of Polyfunctional Arylmagnesium, Arylzinc, and Benzylic Zinc Reagents by Using Magnesium in the Presence of LiCl. Chemistry – A European Journal 2009, 15, 7192-7202, 10.1002/chem.200900575.
- E. J. Corey; A. Venkateswarlu; Protection of hydroxyl groups as tert-butyldimethylsilyl derivatives. Journal of the American Chemical Society 1972, 94, 6190-6191, 10.1021/ja00772a043.
- Shuji Futagawa; Toshishige Inui; Tetsuo Shiba; Nuclear Magnetic Resonance Study of the Stereoisomeric 2-Oxazolidone and 2-Phenyl-2-oxazoline Derivatives of α-Amino-β-hydroxy Acids. Bulletin of the Chemical Society of Japan 1973, 46, 3308-3310, 10.1246/bcsj.46.3308.
- Jekishan R. Parikh; William V. E. Doering; Sulfur trioxide in the oxidation of alcohols by dimethyl sulfoxide. Journal of the American Chemical Society 1967, 89, 5505-5507, 10.1021/ja00997a067.
- Chriss McDonald; Harald Holcomb; Kenneth Kennedy; Elijah Kirkpatrick; Todd Leathers; Penny Vanemon; The N-iodosuccinimide-mediated conversion of aldehydes to methyl esters. The Journal of Organic Chemistry 1989, 54, 1213-1215, 10.1021/jo00266a046.
- Schmidt, U.; Griesser, H.; Leitenberger, V.; Lieberknecht, A.; Mangold, R.; Meyer, R.; Riedl, B.; Diastereoselective formation of (Z)-didehydroamino acid-esters. Synthesis 1992, 1992, 487-490, .
- Ulrich Schmidt; Albrecht Lieberknecht; Uli Kazmaier; Helmut Griesser; Günther Jung; Jörg Metzger; Amino Acids and Peptides; 75. Synthesis of Di- and Trihydroxyamino Acids - Construction of Lipophilic Tripalmitoyldihydroxy-α-amino Acids. Synthesis 1991, 1991, 49-55, 10.1055/s-1991-26378.
- Lavinia Panella; Alicia Marco Aleixandre; Gerlof J. Kruidhof; Jort Robertus; Bernard Feringa; Johannes G. De Vries; Adriaan Minnaard; Enantioselective Rh-Catalyzed Hydrogenation ofN-Formyl Dehydroamino Esters with Monodentate Phosphoramidite Ligands. The Journal of Organic Chemistry 2006, 71, 2026-2036, 10.1021/jo052451d.
- Michel Van Den Berg; Adriaan Minnaard; Ebe P. Schudde; Jan Van Esch; André H. M. De Vries; Johannes G. De Vries; Ben L. Feringa; Highly Enantioselective Rhodium-Catalyzed Hydrogenation with Monodentate Ligands. Journal of the American Chemical Society 2000, 122, 11539-11540, 10.1021/ja002507f.
- Alexander Kiefer; Uli Kazmaier; Synthesis of modified β-methoxyphenylalanines via diazonium chemistry and their incorporation in desoxycyclomarin analogues. Organic & Biomolecular Chemistry 2019, 17, 88-102, 10.1039/c8ob02777c.
- Uli Kazmaier; Christiane Schneider; Stereoselective Synthesis of Unsaturated Polyhydroxylated Amino Acids via Ester Enolate Claisen Rearrangement. Synlett 1996, 1996, 975-977, 10.1055/s-1996-5637.
- Uli Kazmaier; Christiane Schneider; Application of the Asymmetric Chelate Enolate Claisen Rearrangement to the Synthesis of Unsaturated Polyhydroxylated Amino Acids. Synthesis 1998, 1998, 1321-1326, 10.1055/s-1998-6104.
- Uli Kazmaier; Christiane Schneider; Application of the asymmetric chelate-enolate claise rearrangement to the synthesis of 5-epi-isofagomine. Tetrahedron Letters 1998, 39, 817-818, 10.1016/s0040-4039(97)10855-3.
- Christiane Marti; Erick Carreira; Total Synthesis of (−)-Spirotryprostatin B: Synthesis and Related Studies. Journal of the American Chemical Society 2005, 127, 11505-11515, 10.1021/ja0518880.
- Peng Li; Jie-Cheng Xu; 1-Ethyl 2-Halopyridinium Salts, Highly Efficient Coupling Reagents for Hindered Peptide Synthesis both in Solution and the Solid-Phase. Tetrahedron 2000, 56, 8119-8131, 10.1016/s0040-4020(00)00657-8.
- Peng Li; Jie Cheng Xu; 2-Bromo-1-ethyl Pyridinium Tetrafluoroborate (BEP): A Powerful Coupling Reagent forN-Methylated Peptide Synthesis. Chemistry Letters 2000, 29, 204-205, 10.1246/cl.2000.204.
- Philipp Barbie; Uli Kazmaier; Total synthesis of cyclomarins A, C and D, marine cyclic peptides with interesting anti-tuberculosis and anti-malaria activities. Organic & Biomolecular Chemistry 2016, 14, 6036-6054, 10.1039/c6ob00800c.