 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rakesh K Srivastava | + 15822 word(s) | 15822 | 2020-07-16 10:06:13 | | | |
| 2 | Rakesh K Srivastava | + 15822 word(s) | 15822 | 2020-07-16 10:27:42 | | | | |
| 3 | Rakesh K Srivastava | + 453 word(s) | 16275 | 2020-07-16 10:36:12 | | | | |
| 4 | Rakesh K Srivastava | + 453 word(s) | 16275 | 2020-07-16 10:38:36 | | | | |
| 5 | Rakesh K Srivastava | + 453 word(s) | 16275 | 2020-07-16 10:42:00 | | | | |
| 6 | Rakesh K Srivastava | + 48 word(s) | 15870 | 2020-07-16 11:13:27 | | | | |
| 7 | Vicky Zhou | -14173 word(s) | 1697 | 2020-10-29 06:55:32 | | | | |
| 8 | Vicky Zhou | + 1 word(s) | 1698 | 2020-10-29 07:03:01 | | |
Video Upload Options
The phenomenon of heterosis has fascinated plant breeders ever since it was first described by Charles Darwin in 1876 in the vegetable kingdom and later elaborated by George H Shull and Edward M East in maize during 1908. Heterosis is the phenotypic and functional superiority manifested in the F1 crosses over the parents. Various classical complementation mechanisms gave way to the study of the underlying potential cellular and molecular mechanisms responsible for heterosis. In cereals, such as maize, heterosis has been exploited very well, with the development of many single-cross hybrids that revolutionized the yield and productivity enhancements.
1. Introduction
Heterosis (syn hybrid vigor) is a natural phenomenon whereby hybrid (first filial generation, i.e., F1) offsprings of genetically diverse individuals exhibit improved physical and functional features relative to their parents [1][2]. Heterosis has been studied in most eukaryotic organisms, including plants, animals, and fungi. When two homozygous inbred lines (true breeding line derived from recurrent inbreeding) with distinct genetic constitutions are hybridized together, the resultant hybrids have greater height, weight, fertility, robustness, and constitutional vigor than either of the parents and their self-pollinated counterparts [3]. Naturally cross-pollinating species like pearl millet, rye, maize, and other grasses typically exhibit a higher degree of heterosis than the self-pollinating crop plants such as rice, barley, wheat, and oats. Nevertheless, many hybrid cultivars have also been developed in self-pollinating plant species [4]. Heterosis can manifest by virtue of improvement of several traits during plant growth and development. In pearl millet, considerable growth differences between hybrids and their parents can be monitored during different stages of growth and development. Post-germination, two root architectural traits such as primary root length and lateral root density show differences at early as well as later stages. During late development stages, the F1 hybrids display relatively more luxurious growth, greater biomass accumulation, and higher seed setting than their parent genotypes. It is noteworthy that the degree of heterosis can vary considerably among various traits. For example, grain iron (Fe) and zinc (Zn) contents in pearl millet F1 hybrids do not improve over their parental lines and require high levels of micronutrients in both parents. The maximum level of heterosis is monitored in the F1 derived from cross-pollination between diverse genotypes. This demonstrates the involvement of several alleles or genetic loci with diverse interactions at cellular and molecular levels. While the superiority of the progenies over their parents is progressively reduced in the successive generations developed from self-pollination. Heterosis has been exploited in many cereals, oilseeds, vegetables, fruits, pulses, etc., through the development of F1 hybrids globally [4].
2. Molecular Bases of Heterosis
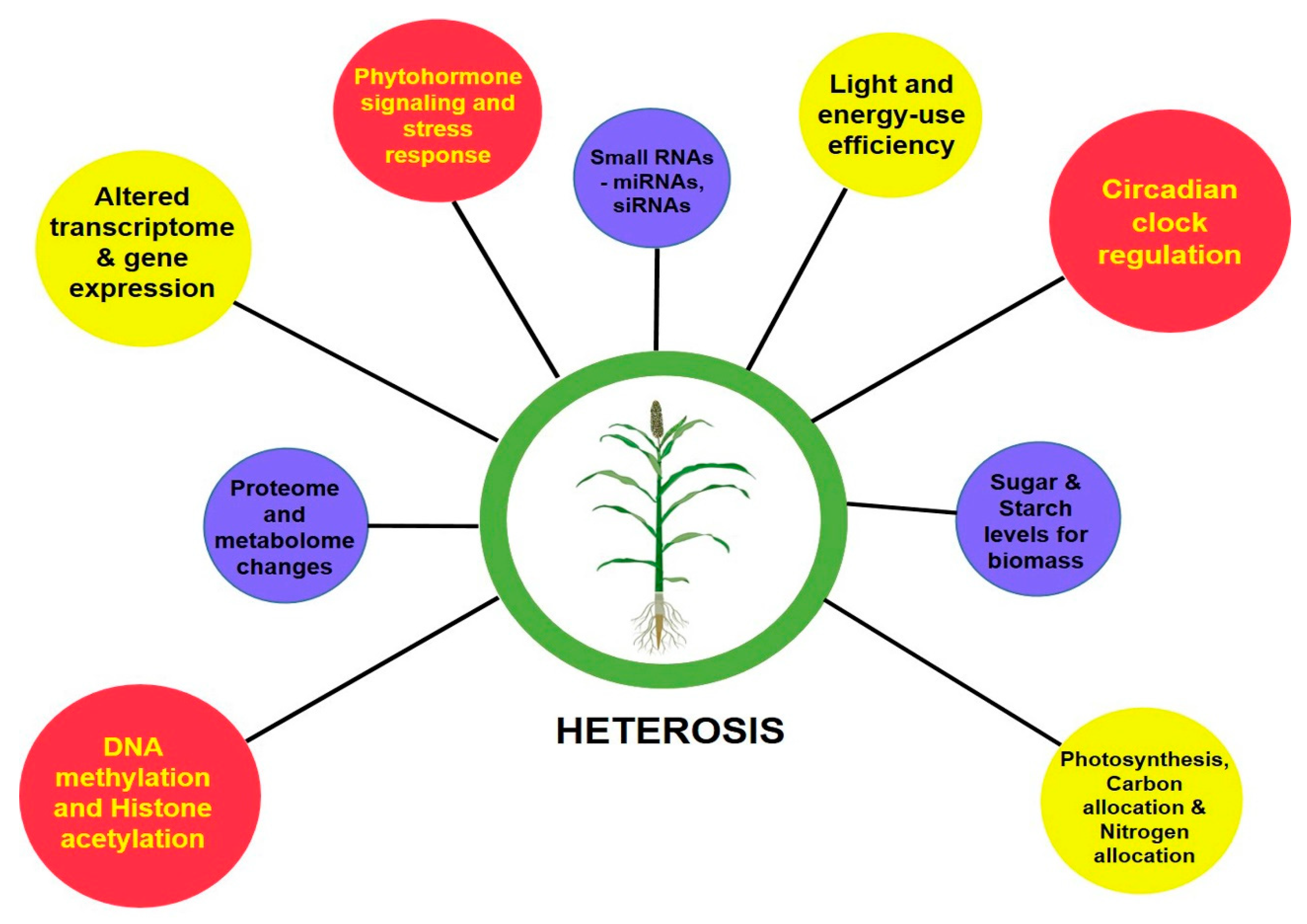
The perceived greater vigor in the heterotic phenotype than its inbred parental lines is the cumulative result of the coding of genetic information by transcription, translation and their levels of regulation. With the advent of next-generation sequencing (NGS) technologies, genome-wide single-nucleotide polymorphisms (SNPs), insertion or deletions (indels), large structural variations in the form of presence and absence variations (PAVs), and copy number variations (CNVs) that contribute to the phenotypic diversity can be easily deciphered [5][6][7]. To decode the underlying mechanisms determining the degree of vigor differences between inbreds and hybrids, molecular studies were advanced to assess their transcriptome, proteome, epigenome and other regulatory related mechanisms [8]. At a glance, Figure 1 gives a basic idea on the molecular bases of heterosis.
2.1. Transcriptomics View on Heterosis
Hybridization of the inbred parental lines leads to interactions between the nucleus and cytoplasm and the resulting changes at the cellular and molecular level leads to the altered patterns of gene expression. Genome-wide modifications in the gene expression levels and their mechanism of actions in the hybrids vis-à-vis its inbreds have been documented in several hybrids of maize [9][10][11], rice [12], wheat [13], cotton [14], Arabidopsis [15][16][17], etc. Transcriptomics and its potential in heterosis can best be viewed as a transitional phase in between the genetic information and the plant phenotype which specifically measures the relative contribution of each allele in a hybrid [18]. To identify the genes involved in heterosis using transcriptome analyses, technologies like microarray, RNA-sequencing (RNA-seq), etc., are being used to compare inbred parental lines with their F1 progenies. From the initial transcriptomic studies on several crop species, it was believed that favorable gene expression levels in hybrids are predominant when compared to its inbred parents [19][20][21][22]. However, it is important to note that differential gene expression levels may not directly correspond to the protein activity between inbred parental lines and hybrids, or to the observed heterosis in the hybrids, and post transcription/translation regulations need to be taken into account [23][24].
2.2. Proteomics View on Heterosis
Studying the role of proteins in determining heterosis is important, as changes at the level of transcripts may not always reflect at the protein level due to various post-transcriptional and translational regulatory mechanisms [24]. The generalized concept is that, in the inbred parental lines protein metabolism is higher due to unstable protein levels which require more energy to quench. Consequently, less energy remains for its vegetative growth, biomass, and yield in the end. The genetic basis for this condition in inbred parental lines is due to the lack of allelic choice in their homozygous condition with only two alleles, whereas hybrids in polyploidy condition will have more alleles and thus display faster growth rates due to increased cell divisions [25].
With the advent of 2-D gel electrophoresis (2-DGE) along with mass spectrometry (MS), numerous studies have been reported to identify the differentially expressed proteins (DEPs) determining heterosis. In addition, MS-based protein detection and quantification depend upon the two isobaric labeling reagents like tandem mass tags (TMT) and isobaric tags for relative and absolute quantification (iTRAQ), which are employed to detect altered proteins or DEPs in heterotic phenotypes [24][26]. To date, the majority of the DEPs determining heterosis have been identified in major crop species like maize, wheat and rice from the tissue samples of the embryo, root, and leaf [27][28][29]. It is apparent from these studies that the majority of the DEPs identified between inbreds and hybrids are due to the non-additive gene effects, and it has also been reported that these DEPs belong to the pathways of signal transduction, glycolysis, photosynthesis, disease resistance, carbon metabolism, protein, amino acid metabolism, etc. [30]. These results indicate that the degree of heterosis was dependent upon the incidence of protein isoforms or modifications [8].
2.3. Epigenomics View on Heterosis
DNA methylation: The resulting vigor in hybrids after the hybridization of two distant inbred parental lines is often linked with epigenetic modifications, viz., DNA methylation [31], chromatin structure modification [32], histone acetylation [33], or small RNA induced regulations, etc. [34]. Genome activity and the cellular development of any crop species are indeed regulated by DNA methylation. In most plant species, DNA methylation takes place on the 5’ position of cytosine residues at CG, CHH and CHG regions (where H may be A, T or C) by DNA methyltransferases [35][36]. The degree of DNA methylation change in the hybrids depends on the diversity among inbred parents [37]. Using bisulfite and siRNA sequencing, differential methylated loci were identified in the rice hybrids (Nipponbare and Indica) and their parents to decipher the role of DNA methylation in causing epigenetic heritability [38]. In the allopolyploids of Arabidopsis, genes with cis- but not trans-regulatory changes were augmented in loci that were hypo-methylated or hyper-methylated [39]. The manifestation of heterosis by DNA methylation is mainly mediated by the suppression of the transcription process of the regulatory genes involved in enhancing inbreeding depression or by promoting the expression of genes for heterosis [40]. Greaves et al. [41] suggested that DNA methylation sites in hybrids were frequently associated with regions that are differentially methylated in their inbred parents. In particular, methylated regions in the inbred parents were usually covered by the siRNA levels, and this implies that RNA-directed DNA methylation (RdDM) pathway may induce the remodeling of DNA methylation sites in hybrids to manifest heterosis [42].
Apart from DNA methylation, another epigenetic system involved in hybrid vigor is histone modification. These modifications can occur at the post-translational level for the amino acids in histone proteins at the N-terminal tails in the form of methylation, phosphorylation, and acetylation of certain residues [43]. Usual modifications will occur with the histones like H3K9ac and H3K4me3 found in euchromatin regions with active gene expression. Meanwhile, histones H3K27me3 and H3K4me3 were found in the pericentromeric, heterochromatin and transposable element (TE) regions with low transcript levels [44]. He et al. [12] compared differential expression patterns of H3K4me3 and H3K27me3 between hybrids and parents of rice subspecies and found that H3K4me3 is transcriptionally active and H3K27me3 is inactive. In maize F1 hybrid endosperm-derived transcriptomes, a histone alternate HTA112 showed significant variations in expression when compared to its parental lines [45]. These studies, though limited, raised the possibility of inducing heterosis through epigenetic histone modifications.
Small RNAs: Small RNAs, mediating gene expression, and epigenetic regulation refer to the class of microRNAs (miRNAs), small interfering RNAs (siRNAs), and trans-acting siRNAs (tasiRNAs) [46]. Usually, siRNAs will maintain the genome stability by affecting the genes tagged with transposable elements (TEs) and also by retaining the stable inheritance of repeat-associated siRNAs, whereas miRNAs and tasiRNAs are involved in controlling morphological and developmental traits [47]. The combination of distant inbred parental siRNAs in the hybrids will exert both cis and trans-acting effects on TEs and TE-coding genes which causes genomic instability thereby leading to the abortive embryo and endosperm formations termed as hybrid lethality [48]; on the contrary, some small RNAs also play positive regulatory roles in terms of protection from the genomic shock, ultimately leading to hybrid vigor. Small RNA gene expression studies in the hybrids of rice, Arabidopsis and maize revealed the downregulated levels of 24-nt siRNAs when compared to their inbred parental lines. The 24-nt siRNAs that are involved in the RdDM mechanism will finally lead to transcriptional repression by stimulating the DNA methylation process. Hence, the reduction of these siRNA levels may lead to the ample expression of protein-coding genes in hybrids [49][50]. The differential expression of siRNAs in hybrids relative to their parents is due to the differences in the promoter regions of small RNA coding genes. Therefore, the epigenetically developed differences at the gene expression level of 24-nt siRNAs in hybrids may contribute to heterosis [51][12][52].
References
- Coors, C.G.; Pandey, S. (Eds.) The Genetics and Exploitation of Heterosis in Crops; American Society of Agronomy: Madison, WI, USA, 1999; pp. 99–118.
- Shull, G.H. What is heterosis? Genetics 1948, 33, 439–446.
- Darwin, C.R. The Effects of Cross and Self Fertilization in the Vegetable Kingdom; John Murray: London, UK, 1876; p. 482.
- Rajendrakumar, P.; Hariprasanna, K.; Seetharama, N. Prediction of Heterosis in Crop Plants—Status and Prospects. Am. J. Exp. Agric. 2015, 9, 1–16.
- Díaz, A.; Zikhali, M.; Turner, A.S.; Isaac, P.; Laurie, D.A. Copy number variation affecting the photoperiod-B1 and vernalization-A1 genes is associated with altered flowering time in wheat (Triticum aestivum). PLoS ONE 2012, 7, 1–11.
- Zmienko, A.; Samelak-Czajka, A.; Kozlowski, P.; Figlerowicz, M. Copy number polymorphism in plant genomes. Theor. Appl. Genet. 2013, 127, 1–18.
- Saxena, R.K.; Edwards, D.; Varshney, R.K. Structural variations in plant genomes. Brief. Funct. Genom. 2014, 13, 296–307.
- Kaeppler, S. Heterosis: Many Genes, Many Mechanisms—End the Search for an Undiscovered Unifying Theory. Isrn Bot. 2012, 2012, 1–12.
- Swanson-Wagner, R.A.; Jia, Y.; DeCook, R.; Borsuk, L.A.; Nettleton, D.S.; Schnable, P.S. All possible modes of gene action are observed in a global comparison of gene expression in a maize F1 hybrid and its inbred parents. Proc. Natl. Acad. Sci. USA 2006, 103, 6805–6810.
- Guo, M.; Rupe, M.A.; Yang, X.; Crasta, O.; Zinselmeier, C.; Smith, O.S.; Bowen, B. Genome-wide transcript analysis of maize hybrids: Allelic additive gene expression and yield heterosis. Theor. Appl. Genet. 2006, 113, 831–845.
- Stupar, R.M.; Springer, N.M. Cis-transcriptional Variation in Maize Inbred Lines B73 and Mo17 Leads to Additive Expression Patterns in the F1Hybrid. Genetics 2006, 173, 2199–2210.
- He, G.; Zhu, X.; Elling, A.A.; Chen, L.; Wang, X.; Guo, L.; Liang, M.; He, H.; Zhang, H.; Chen, F.; et al. Global Epigenetic and Transcriptional Trends among Two Rice Subspecies and Their Reciprocal Hybrids. Plant. Cell 2010, 22, 17–33.
- Wang, Z.; Ni, Z.; Wu, H.; Nie, X.; Sun, Q. Heterosis in root development and differential gene expression between hybrids and their parental inbreds in wheat (Triticum aestivum L.). Theor. Appl. Genet. 2006, 113, 1283–1294.
- Flagel, L.; Udall, J.A.; Nettleton, D.S.; Wendel, J.F. Duplicate gene expression in allopolyploid Gossypium reveals two temporally distinct phases of expression evolution. BMC Biol. 2008, 6, 16.
- Shen, H.; He, H.; Li, J.; Chen, W.; Wang, X.; Guo, L.; Peng, Z.; He, G.; Zhong, S.; Qi, Y.; et al. Genome-Wide Analysis of DNA Methylation and Gene Expression Changes in Two Arabidopsis Ecotypes and Their Reciprocal Hybrids. Plant. Cell 2012, 24, 875–892.
- Fujimoto, R.; Taylor, J.; Shirasawa, S.; Peacock, W.J.; Dennis, E.S. Heterosis of Arabidopsis hybrids between C24 and Col is associated with increased photosynthesis capacity. Proc. Natl. Acad. Sci. USA 2012, 109, 7109–7114.
- Fujimoto, R; Taylor, J.M; Sasaki, T; Kawanabe, T; Dennis, E.S. Genome wide gene expression in artificially synthesized amphidiploids of Arabidopsis. Plant Mol. Biol. 2011, 77, 419–431.
- Schnable, P.S.; Springer, N.M. Progress Toward Understanding Heterosis in Crop Plants. Annu. Rev. Plant. Biol. 2013, 64, 71–88.
- Comings, D.E.; MacMurray, J.P. Molecular Heterosis: A Review. Mol. Genet. Metab. 2000, 71, 19–31.
- Baranwal, V.K.; Mikkilineni, V.; Zehr, U.B.; Tyagi, A.K.; Kapoor, S. Heterosis: Emerging ideas about hybrid vigour. J. Exp. Bot. 2012, 63, 6309–6314.
- Stupar, R.M.; Gardiner, J.; Oldre, A.; Haun, W.J.; Chandler, V.L.; Springer, N.M. Gene expression analyses in maize inbreds and hybrids with varying levels of heterosis. BMC Plant. Biol. 2008, 8, 33.
- Fujimoto, R.; Uezono, K.; Ishikura, S.; Osabe, K.; Peacock, W.J.; Dennis, E.S. Recent research on the mechanism of heterosis is important for crop and vegetable breeding systems. Breed. Sci. 2018, 68, 145–158.
- Fu, D.; Xiao, M.; Hayward, A.; Fu, Y.; Liu, G.; Jiang, G.; Zhang, H. Utilization of crop heterosis: A review. Euphytica 2014, 197, 161–173.
- Xing, J.; Sun, Q.; Ni, Z. Proteomic patterns associated with heterosis. Biochim. Biophys. Acta (Bba)—Proteins Proteom. 2016, 1864, 908–915.
- Goff, S.A. A unifying theory for general multigenic heterosis: Energy efficiency, protein metabolism, and implications for molecular breeding. New Phytol. 2010, 189, 923–937.
- Wang, J.; Yu, Q.; Xiong, H.; Wang, J.; Chen, S.; Yang, Z.; Dai, S. Proteomic Insight into the Response of Arabidopsis Chloroplasts to Darkness. PLoS ONE 2016, 11, e0154235.
- Guo, B.; Chen, Y.; Zhang, G.; Xing, J.; Hu, Z.; Feng, W.; Yao, Y.; Peng, H.; Du, J.; Zhang, Y.; et al. Comparative Proteomic Analysis of Embryos between a Maize Hybrid and Its Parental Lines during Early Stages of Seed Germination. PLoS ONE 2013, 8, e65867.
- Song, X.; Ni, Z.; Yao, Y.; Xie, C.; Li, Z.; Wu, H.; Zhang, Y.; Sun, Q. Wheat (Triticum aestivum L.) root proteome and differentially expressed root proteins between hybrid and parents. Proteomics 2007, 7, 3538–3557.
- Zhang, C.; Yin, Y.; Zhang, A.; Lu, Q.; Wen, X.; Zhu, Z.; Zhang, L.; Lu, C. Comparative proteomic study reveals dynamic proteome changes between super hybrid rice LYP9 and its parents at different developmental stages. J. Plant. Physiol. 2012, 169, 387–398.
- Marcon, C.; Schutzenmeister, A.; Schutz, W.; Madlung, J.; Piepho, H.P.; Hochholdinger, F. Non-additive protein accumulation patterns in maize (Zea mays L) hybrids during embryo development. J. Prot. Res. 2010, 9, 6511–6522.
- Parisod, C.; Salmon, A.; Zerjal, T.; Tenaillon, M.; Grandbastien, M.-A.; Ainouche, M. Rapid structural and epigenetic reorganization near transposable elements in hybrid and allopolyploid genomes in Spartina. New Phytol. 2009, 184, 1003–1015.
- Moghaddam, A.M.B.; Colot, V.; Mette, F.; Houben, A. Heterosis and chromatin structure: Does intraspecific hybridization trigger epigenetic changes? Chrom. Res. 2007, 15, 23.
- Tanabata, T.; Taguchi-Shiobara, F.; Kishimoto, N.; Chechetka, S.; Shinomura, T.; Habu, Y. A phenomics approach detected differential epigenetic growth regulation between inbreds and their hybrid in Oryza sativa. Mol. Breed. 2010, 26, 729–734.
- Groszmann, M.; Greaves, I.K.; Albert, N.; Fujimoto, R.; Helliwell, C.A.; Dennis, E.S.; Peacock, W.J. Epigenetics in plants—Vernalization and hybrid vigour. Biochim. Biophys. Acta 2011, 1809, 427–437.
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220.
- Fernie, A.R.; Chen, B.; Wang, X.; Li, X.; Li, J.; He, H.; Yang, M.; Lu, L.; Qi, Y.; Wang, X.; et al. Conservation and divergence of transcriptomic and epigenomic variation in maize hybrids. Genome Biol. 2013, 14, R57.
- Chen, Z.J. Genomic and epigenetic insights into themolecular bases of heterosis. Nat. Rev. Genet. 2013, 14, 471–482.
- Chodavarapu, R.K.; Feng, S.; Ding, B.; Simon, S.A.; Lopez, D.; Jia, Y.; Wang, G.-L.; Meyers, B.C.; Jacobsen, S.E.; Pellegrini, M. Transcriptome and methylome interactions in rice hybrids. Proc. Natl. Acad. Sci. USA 2012, 109, 12040–12045.
- Shi, X.; Ng, D.W.-K.; Zhang, C.; Comai, L.; Ye, W.; Chen, Z.J. Cis- and trans-regulatory divergence between progenitor species determines gene-expression novelty in Arabidopsis allopolyploids. Nat. Commun. 2012, 3, 950.
- Nakamura, S.; Hosaka, K. DNA methylation in diploid inbred lines of potatoes and its possible role in the regulation of heterosis. Theor. Appl. Genet. 2009, 120, 205–214.
- Greaves, I.K.; Groszmann, M.; Ying, H.; Taylor, J.; Peacock, W.J.; Dennis, E.S. Trans chromosomal methylation in Arabidopsis hybrids. Proc. Natl. Acad. Sci. USA 2012, 109, 3570–3575.
- Greaves, I.K.; Eichten, S.R.; Groszmann, M.; Wang, A.; Ying, H.; Peacock, W.J.; Dennis, E.S. Twenty-four–nucleotide siRNAs produce heritable trans-chromosomal methylation in F1 Arabidopsis hybrids. Proc. Natl. Acad. Sci. USA 2016, 113, E6895–E6902.
- Berger, S.L. The complex language of chromatin regulation during transcription. Nature 2007, 447, 407–412.
- Roudier, F.; Teixeira, F.K.; Colot, V. Chromatin indexing in Arabidopsis: An epigenomic tale of tails and more. Trends Genet. 2009, 25, 511–517.
- Jahnke, S.; Sarholz, B.; Thiemann, A.; Kühr, V.; Gutierrez-Marcos, J.F.; Geiger, H.H.; Piepho, H.-P.; Scholten, S. Heterosis in early seed development: A comparative study of F1 embryo and endosperm tissues 6 days after fertilization. Theor. Appl. Genet. 2009, 120, 389–400.
- Chen, X. Small RNAs and their roles in plant development. Annu. Rev. Cell Dev. Biol. 2009, 25, 21–44.
- Xie, F.; He, Z.; Esguerra, M.Q.; Qiu, F.; Ramanathan, V. Determination of heterotic groups for tropical Indica hybrid rice germplasm. Theor. Appl. Genet. 2013, 127, 407–417.
- Ng, D.W.-K.; Lu, J.; Chen, Z.J. Big roles for small RNAs in polyploidy, hybrid vigor, and hybrid incompatibility. Curr. Opin. Plant. Biol. 2012, 15, 154–161.
- Greaves, I.K.; Gonzalez-Bayon, R.; Wang, L.; Zhu, A.; Liu, P.-C.; Groszmann, M.; Peacock, W.J.; Dennis, E.S. Epigenetic Changes in Hybrids. Plant. Physiol. 2015, 168, 1197–1205.
- Groszmann, M.; Greaves, I.K.; Fujimoto, R.; Peacock, W.J.; Dennis, E.S. The role of epigenetics in hybrid vigour. Trends Genet. 2013, 29, 684–690.
- Groszmann, M.; Greaves, I.K.; Albertyn, Z.I.; Scofield, G.N.; Peacock, W.J.; Dennis, E.S. Changes in 24-nt siRNA levels in Arabidopsis hybrids suggest an epigenetic contribution to hybrid vigor. Proc. Natl. Acad. Sci. USA 2011, 108, 2617–2622.
- Barber, W.T.; Zhang, W.; Win, H.; Varala, K.; Dorweiler, J.E.; Hudson, M.E.; Moose, S.P. Repeat associated small RNAs vary among parents and following hybridization in maize. Proc. Natl. Acad. Sci. USA 2012, 109, 10444–10449.


