Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Olivier Provot | + 2065 word(s) | 2065 | 2021-08-09 03:47:45 | | | |
| 2 | Peter Tang | + 24 word(s) | 2089 | 2021-08-10 10:40:25 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Provot, O. Pyrazino[1,2-a]Indoles. Encyclopedia. Available online: https://encyclopedia.pub/entry/12989 (accessed on 27 July 2026).
Provot O. Pyrazino[1,2-a]Indoles. Encyclopedia. Available at: https://encyclopedia.pub/entry/12989. Accessed July 27, 2026.
Provot, Olivier. "Pyrazino[1,2-a]Indoles" Encyclopedia, https://encyclopedia.pub/entry/12989 (accessed July 27, 2026).
Provot, O. (2021, August 10). Pyrazino[1,2-a]Indoles. In Encyclopedia. https://encyclopedia.pub/entry/12989
Provot, Olivier. "Pyrazino[1,2-a]Indoles." Encyclopedia. Web. 10 August, 2021.
Copy Citation
The pyrazino[1,2-a]indole unit is a tricyclic aromatic nucleus combining an indole and a pyrazine linked by the N5 and C9a atoms. The Synthesis and Biological Activities of Pyrazino[1,2-a]Indole and Pyrazino[1,2-a]Indol-1-One Derivatives is presented.
pyrazinoindole
pyrazinoindolone
cyclization
catalysis
biological activity
1. Introduction
The pyrazino[1,2-a]indole unit is a tricyclic aromatic nucleus combining an indole and a pyrazine linked by the N5 and C9a atoms (Figure 1).
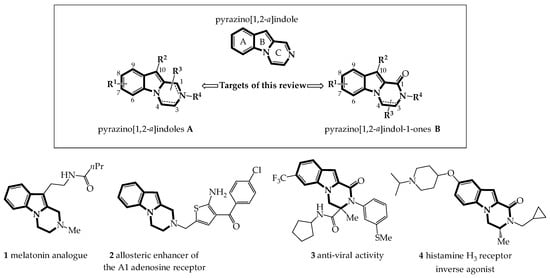
Figure 1. Targets of this review and selection of biologically active tetrahydro-pyrazino[1,2-a]indoles 1a, 2a and dihydro-pyrazino[1,2-a]indol-1-ones 3a, 4a.
The access to this substituted aromatic nucleus has been well studied since 1997 by the chemist community from a synthetic point of view and for its potential in medicinal chemistry [1][2]. In parallel, structural modifications of the pyrazino[1,2-a]indole nucleus showed that (3,4-dihydro)pyrazino[1,2-a]indoles (type A) and (3,4-dihydro)-pyrazino[1,2-a]indol-1-ones (type B) were efficient pharmacophores used in a variety of diseases. To illustrate, 3,4-dihydropyrazinoindoles 1 [3] and 2 [4] (type A) have been showed to be effective at melatonin and adenosine receptors, while 3,4-dihydropyrazinoindol-1-ones 3 [5] and 4 [6] (type B) have been studied for their anti-viral and anti-allergenic activities, respectively.
2. Recent Synthetic Approaches to Variously Substituted Pyrazinoindoles and 3,4-Dihydropyrazinoindoles
The creation of the pyrazino[1,2-a]indole nucleus was mainly achieved by cyclizing indole having various groups (CHO, ketone, imine, nitrile, etc.) on C2 with a nucleophile linked to the indole nitrogen atom, thus creating the pyrazino C-ring (Scheme 1). For example, 2-substituted-1-(prop-2-yn-1-yl)-1H-indoles 5, 7, 9, 11, 13 transformed into pyrazinoindoles 6, 8, 10, 12 and 14 respectively by intramolecular cyclization using NH3 in MeOH, [7][8] DBU under microwave irradiation, [9] AuCl3 [10] as triple bond activator, Ni(OAc)2 in the presence of hydroxylamine [11] or NaH in DMF [12].
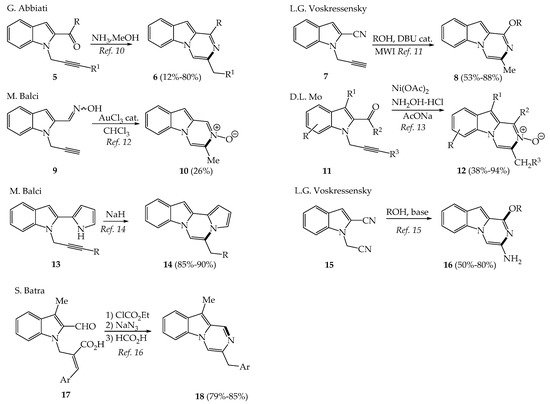
Scheme 1. Synthesis of pyrazinoindoles.
The C-ring of the pyrazino[1,2-a]indole system has been also built by alcoholate promoted cyclization of indolodinitrile compound 15 [13] and by Curtius reaction using Morita–Baylis–Hillman derivatives 17 [14] with good to excellent yields.
The synthesis of variously substituted pyrazinoindoles having a saturated C-ring has been more studied than that of their aromatic counterparts, probably because these compounds offer more functional diversity such as diastereoselective accesses, but mainly because they have shown superior efficacy in medicinal chemistry. Among the simplest reactions described to prepare these compounds was the one proposed by Katritzky, who used a cycloaddition reaction between a N-ethylamine-indole 19 and formaldehyde in the presence of benzotriazole (Bt) (Scheme 2). A subsequent nucleophilic substitution reaction of the benzotriazole gives rise to various N-substituted pyrazinoindoles 20 [15]. N-ethylamine-indoles 19 also reacted, in a complementary approach, with aldehydes and Bt in the presence of Lewis acids to give C1-substituted pyrazinoindoles 21 [16].
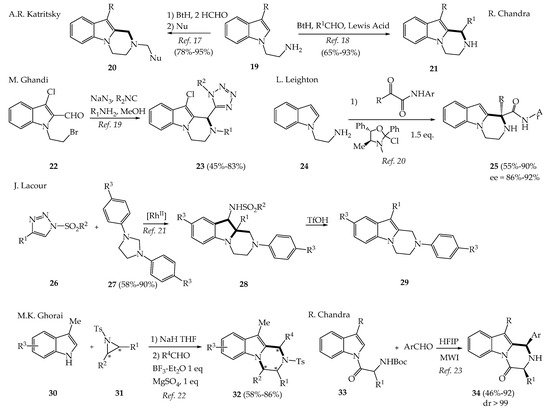
Scheme 2. Synthesis of 1,2,3,4-tetrahydro-pyrazinoindoles.
A Ugi-azide four component approach was recently published to prepare a series of N-substituted pyrazinoindoles 23 having on C1 a substituted tetrazole ring [17]. Leighton et al. proposed highly enantioselective iso-Pictet–Spengler reactions using the condensation of 2-(1H-indol-1-yl)ethanamine 24 with a variety of α-ketoamides, followed by the addition of a commercially available chiral silicon Lewis acid (L*) to give 1,1-disubstituted-tetrahydropyrazino[1,2-a]indoles 25 with good yields (55–90%) and high enantioselectivity (ee = 86–96%) [18]. Guinchard et al. also reported an Au(I)-catalyzed Pictet–Spengler reaction to prepare a variety of complex heterocyclic compounds including tetrahydro-pyrazinoindoles with good yields ranging from 43 to 93% [19]. In 2021, Lacour et al. reported that N-sulfonyltriazoles 26 and imidazolines 27 reacted under rhodium catalysis to give a variety of hexahydro-pyrazinoindoles 28 with excellent yields easily transformed in tetrahydropyrazinoindoles 29 after a welcome rearrangement in triflic acid TfOH [20]. Ghorai et al. reported in 2018 of an elegant synthesis of 1,3-disubstituted 1,2,3,4-tetrahydropyrazino[1,2-a]indoles 32 with excellent stereoselectivity (de, ee >99%) via base-mediated ring opening of chiral aziridines 31 with skatoles 30 followed by BF3-OEt2 catalyzed Pictet–Spengler reaction [21]. Chandra group reported synthesis of di-substituted pyrazinoindol-4-ones 34 with an excellent diastereoselectivity (>99%) via a Pictet–Spengler reaction by mixing 3-substituted-N-acylindoles 33 and aromatic aldehydes in the presence of hexafluoroisopropanol (HFIP) under microwave irradiation [22].
3. Biologically Active Pyrazino[1,2-a]indoles
3.1. Neuropsychiatric Properties
Bos et al., in a program dedicated to the discovery of novel drugs for the treatment of neuropsychiatric disorders, synthesized a variety of pyrazino[1,2-a]indoles 36a–f which were found as partial agonist ligands at the 5HT2C receptor (Scheme 3) [23].
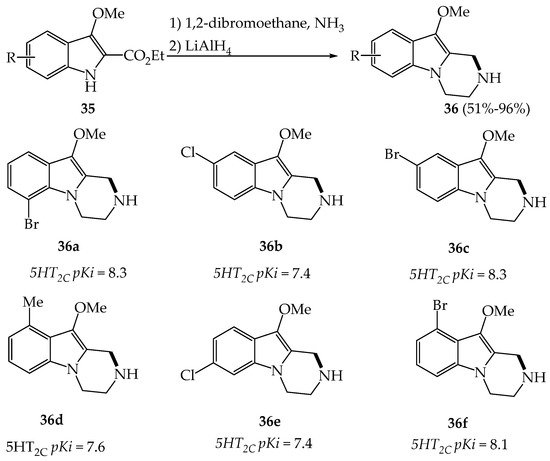
Scheme 3. Synthesis of tetrahydro-pyrazinoindoles 36 and their binding data at the 5HT2C receptor subtype.
Pyrazinoindole derivatives 36a–f were prepared according to a N-alkylation/cyclization/reduction sequence of indoles 35 having an ester function on C2 [23]. Pyrazinoindoles 36 were found to be partial agonists at the 5HT2C receptor subtype binding with a higher affinity than for 5HT2A receptors. Best affinities for 5HT2C receptor were observed for 10-methoxy-pyrazinoindoles having on 6, 7, 8 or 9-position of the A-ring bulky atoms (F < Me < Cl < Br). In animals, 36d showed a 30-fold selectivity for 5HT2C receptors compared to 5HT2A receptors and an only 3-fold selectivity compared to 5HT1A receptors. In vivo results (rats and monkeys) also demonstrated that pyrazinoindole 36d had a promising therapeutic potential for the treatment of various psychiatric disorders, such as obsessive-compulsive disorders, panic anxiety or depression.
Imidazoline receptors exist in two forms, I1 and I2, for which there are very few ligands that are selective for one of the two forms. As a result, it is very difficult to assign a well-defined role to them even though I2 receptors have been described as involved in a variety of CNS disorders. Tetrahydro-pyrazinoindoles 37a–c were evaluated by Glennon group for their potential as I2 imidazoline receptor ligands [24] due to their resemblance to β-carbolines [25] and imidazo-pyridoindoles (Figure 2) [26].
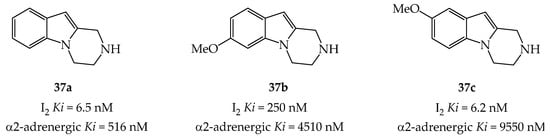
Figure 2. Tetrahydro-pyrazinoindoles 37 and their binding data at I2 and α-adrenergic receptors.
Remarkably, 8-methoxypyrazinoindole 37c binds to I2 receptors with high affinity (Ki = 6.2 nM) and has a 1500-fold selectivity for I2 receptors compared to α2-adrenergic receptors (Ki = 9550 nM) and a 1000-fold selectivity for I1 receptors. A similar high selectivity for 37c was also observed towards I2 receptors compared to serotonin 5HT2A and 5HT2C receptors.
Zlotos et al. synthesized a series of C1-substituted tetrahydro-pyrazinoindoles 1 and 39 as novel potent melatoninergic ligands from 38 in 4 steps (Scheme 4) [3].
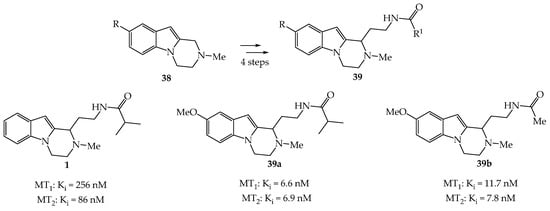
Scheme 4. Synthesis of tetrahydro-pyrazinoindoles 1 and 39.
The affinity of pyrazinoindoles 1, 39a,b for human MT1 and MT2 melatonin receptors in Chinese Hamster Ovary (CHO) cells was measured by competition binding analysis using 2-[125I]-iodomelatonin. The most active compound 39a was found to be an interesting ligand for MT1 and MT2 receptors with excellent affinity, but with no subtype selectivity (MT1: Ki = 6.6 nM; MT2: Ki = 6.9 nM, respectively). This tetrahydro-pyrazinoindole compound was found to be a partial agonist at MT1 receptors and possessed no intrinsic activity at MT2 receptors. It is noteworthy that the treatment of pyrazinoindole 7 (R = H) with MeI gave the corresponding N-dimethyliodonium salt which was found to displace [3H]-cytisine from the nicotinic binding sites on rat cerebral cortex and was revealed to be a nicotinic agonist ligand [27].
3.2. Auto-Immune Properties
Among the C1-substituted pyrazinoindoles, we can cite the work of Buzard et al. who prepared a series of C3-tetrahydro-pyrazinoindoles 42 from the same precursor 41 resulting from an intramolecular Michael reaction carried out on mesylate 40 in the presence of NH3 (Scheme 5) [28]. In a previous work, Buzard et al. showed that some cyclopenta[b]indoles were very potent agonists of the sphingosine 1-phosphate (S1P1) receptor that could be used for the treatment of certain autoimmune diseases [29]. Due to the structural resemblance to these indoles, a series of tricyclic analogues (pyridoindoles, oxazinoindoles and pyrazinoindoles) were designed, synthesized, and evaluated. Pyridoindoles proved to be the most promising compounds in this series of fused-indole compounds, even if pyrazinoindoles 42a–d, prepared from 41 in four steps (N-Boc protection, O-debenzylation, O-functionalization with various benzyl chlorides and t-Butylester hydrolysis) showed interesting activities as S1P1 receptor agonists with nanomolar EC50 values. For the treatment of autoimmune disease as rheumatoid arthritis, Hill et al. synthesized a pyrazinoindole derivative having on C10 a substituted maleimide nucleus which was unfortunately found to be poorly active as protein kinase C inhibitor (IC50 = 540 nM) [30].
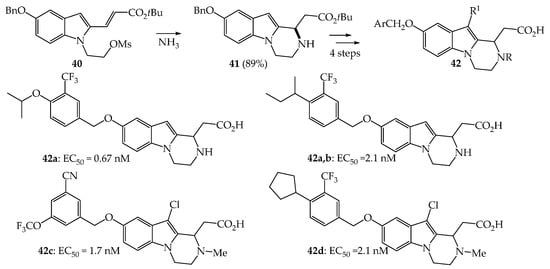
Scheme 5. Synthesis of tetrahydro-pyrazinoindole 41 and human S1P1 cAMP EC50 values of derivatives 42a–d.
3.3. Anti-Bacterial and Anti-Fungal Properties
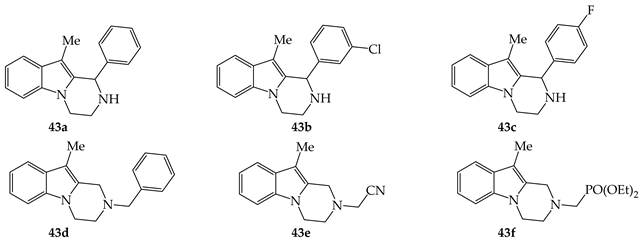
A series of 15 pyrazinoindoles 43 were prepared according to Refs. [15][16] (see Scheme 2) by Verma group and evaluated for their anti-bacterial properties (Table 1) [31]. The in vitro antibacterial activity was evaluated by disc diffusion assay (DDA) using pathogenic strains of Staphylococcus aureus, Salmonella typhi, Streptomyces thermonitrificans, Pseudomonas aeruginosa and Escherichia coli. It was demonstrated that 43a was only active on P. aeruginosa and, similarly, a significant activity on P. aeruginosa and S. thermonitrificans was noticed with 43b. Pyrazinoindoles 43c–e were found to be active against all tested strains but with a relatively modest efficacy when compared with gentamycin. From these results, it seems that the presence of substituents on the nitrogen atom of pyrazinoindoles is deleterious for a satisfactory anti-bacterial activity. Pyrazinoindoles having an aromatic C-ring were not active against all tested strains.
Table 1. Anti-bacterial properties of tetrahydro-pyrazinoindoles 43a–f.
|
Cpnd |
DDA Minimum Inhibitory Concentrations (μg/disc) |
|||
|---|---|---|---|---|
|
S. aureus |
S. typhi |
P. aeruginosa |
S. thermonitrificans |
|
|
43a |
- |
- |
3.75 |
- |
|
43b |
- |
- |
15 |
3.75 |
|
43c |
30 |
30 |
30 |
7.5 |
|
43d |
15 |
60 |
60 |
60 |
|
43e |
15 |
30 |
60 |
30 |
|
43f |
- |
- |
60 |
60 |
|
Gentamycin |
1 |
1 |
0.5 |
1 |
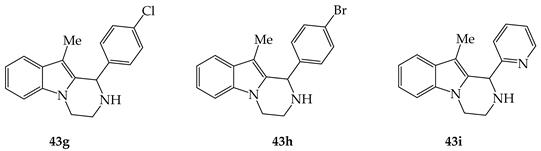
Tetrahydro-pyrazinoindoles 43 were also evaluated for their anti-fungal activity against Aspergillus flavus, Aspergillus fumigatus, Aspergillus. niger and Candida albicans (Table 2) [32]. The anti-Aspergillus activity was evaluated by disc diffusion assay (DDA) and the anti-Candida activity was investigated by microbroth dilution assay. The more active tetrahydro-pyrazinoindoles 43 presented in Table 2 displayed a mild to moderate anti-fungal activity, even if these pyrazinoindoles were found to be, in vitro, less cytotoxic than Amphotericin B when used at high concentrations. SARs with compounds 43 were similar for both anti-bacterial and anti-fungal activities.
Table 2. Anti-fungal properties of tetrahydro-pyrazinoindoles 43g–I and 43c.
|
Cpnd |
DDA Minimum Inhibitory Concentrations (μg/disc) |
|||
|---|---|---|---|---|
|
A. flavus |
A. fumigatus |
A. niger |
C. albicans |
|
|
43g |
11.7 |
5.8 |
11.7 |
15.6 |
|
43h |
47 |
23 |
47 |
62.5 |
|
43i |
187 |
94 |
187 |
125 |
|
43c |
47 |
47 |
47 |
125 |
|
Gentamycin |
1 |
1 |
0.5 |
1 |
3.4. Anti-Arrhythmic, Anti-Lipolytic, Neuro- and Cardio-Protective Properties
In a program dedicated to the discovery of ligands able to activate the A1AR adenosine receptor, Romagnoli et al. proposed some derivatizations on PD81,723, an allosteric modulator acting at the A1AR receptor, enhancing the functional effects of adenosine receptor subtype (Scheme 6) [4].
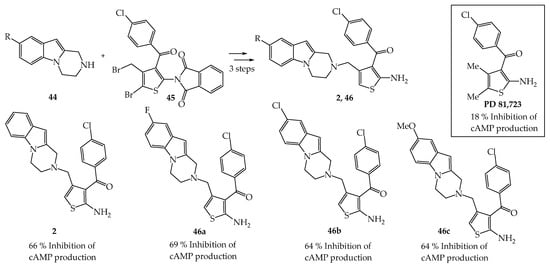
Scheme 6. Synthesis of tetrahydro-pyrazinoindoles 2, 46a–c and effect in cAMP assay in hA1CHO cells.
Thus, pyrazinoindoles 2, 46a–d were synthesized from dibromothiophene 45 and 8-substituted pyrazinoindoles 44 in 3 steps (SN2 reaction, debromination, phthalimide hydrolysis). Pyrazinoindoles 2, 46a–c were next evaluated in a functional assay for their ability to inhibit forskolin stimulated cAMP production via the hA1-AR in intact Chinese hamster ovary (CHO) cells. The four pyrazinoindoles 2, 46a–c were found to be significantly more active than the reference PD 81,723. The best compound 8-fluorated pyrazinoindole 46a inhibited the percentage of cAMP production by 69% vs. 18% for PD 81,723. It was also shown that these derivatives significantly inhibited antagonist binding at the hA1AR, hA2AR or hA3AR receptors.
3.5. Anti-Cancer Properties
Romagnoli et al. studied in 2009 the antiproliferative properties of a series of pyrazinoindoles 17 which were prepared from the reduction/cyclization of N-cyanomethyl derivatives 47 followed by an oxidation reaction using MnO2 (Scheme 7) [33].
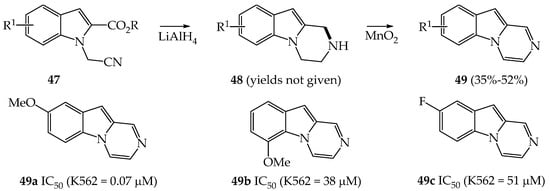
Scheme 7. Synthesis and anti-cancer properties of a selection of pyrazinoindoles 49a–c.
It was shown that pyrazinoindole 49a was the more cytotoxic derivative against human leukemia K562 cancer cells with a promising IC50 value of 0.07 μM. However, this strong cytotoxicity was not observed in other cell lines such as murine leukemia (L1210), murine mammary carcinoma (FM3A), human T-lymphoblastoid (Molt/4 and CEM) and human cervical carcinoma (HeLa) cells with IC50 values superior to 20 μM.
In view of preparing pyrazinoindoles 51 as anti-cancer agents, Kumar et al. mixed N-propargyl indoles 50 having an aldehyde function on C2 with (2-aminophenyl)methanol derivatives in the presence of a catalytic amount of AgNO3 (Scheme 8) [34]. After the reaction of δ-alkynyl aldehydes and nucleophilic anilines, the alcohol function adds on the imine thus creating a second bond (C-O). The third bond creation (C-N) of this process occurs with the nitrogen atom of the imine which reacts with the alkyne triple bond activated by AgNO3 in a 6-exo-dig manner (76–84%).
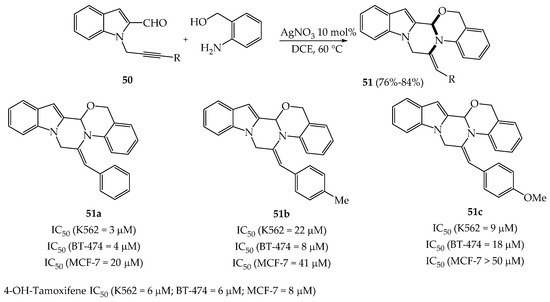
Scheme 8. Synthesis of pyrazinoindoles 51 and their IC50 values against three human cancer cell lines.
Pyrazinoindoles 51 were next evaluated against 3 cancer cell lines (K562 leukemia cells, BT-474 human breast cells; MCF-7 breast cancer cells). As it can be seen in Scheme 8, the more cytotoxic compound was 51a against K562 and BT-474 cancer cells. This pyrazinoindole was significantly more active than 4OH-tamoxifene, used as reference compound, against K562 and BT-474 cells but displayed a lower IC50 value against MCF-7 cancer cells. This result is interesting as 51a exhibited maximum cytotoxicity in p53-deficient cell lines K562 and BT-474 cells but not in p53 wildtype MCF-7 cells. It would certainly be interesting to perform SARs on these structures and to evaluate them on a panel of human cancer lines resistant to the usual treatments.
References
- Singh, A.; Mahapatra, S.; Sewariya, S.; Singh, N.; Singh, S.; Kumar, Y.; Bandichhor, R.; Chandra, R. A mini-review on the synthesis of pyrazinoindole: Recent progress and perspectives. Mini Rev. Org. Chem. 2021, 18, 504–514.
- Sokolova, E.A.; Festa, A.A. Synthesis of pyrazino[1,2-a] indoles and indolo [1,2-a] quinoxalines (microreview). Chem. Heterocycl. Comp. 2016, 52, 219–221.
- Markl, C.; Attia, M.I.; Julius, J.; Sehti, S.; Witt-Enderby, P.A.; Zlotos, D.P. Synthesis and pharmacological evaluation of 1,2,3,4-tetrahydropyrazino[1,2-a] indole and 2-[(phenylmethylamino)methyl]-1H-indole analogues as novel melatoninergic ligands. Bioorg. Med. Chem. 2009, 17, 4583–4594.
- Romagnoli, R.; Baraldi, P.G.; Carrion, M.D.; Cara, C.L.; Salvador, M.K.; Preti, D.; Tabrizi, M.A.; Moorman, A.R.; Vincenzi, F.; Borea, P.A.; et al. Synthesis and biological effects of novel 2-amino-3-(4-chlorobenzoyl)-4-substituted thiophenes as allosteric enhancers of the A1 adenosine receptor. Eur. J. Med. Chem. 2013, 67, 409–427.
- Kounde, C.S.; Yeo, H.Q.; Wang, Q.Y.; Wan, K.F.; Dong, H.; Karuna, R.; Dix, I.; Wagner, T.; Zou, B.; Simon, O. Discovery of 2-oxopiperazine dengue inhibitors by scaffold morphing of a phenotypic high-throughput screening hit. Bioorg. Med. Chem. Lett. 2017, 27, 1385–1389.
- Richter, H.G.F.; Freichel, C.; Huwyler, J.; Nakagawa, T.; Nettekoven, M.; Plancher, J.-M.; Raab, S.; Roche, O.; Schuler, F.; Taylor, S.; et al. Discovery of potent and selective histamine H3 receptor inverse agonists based on the 3,4-dihydro-2H-pyrazino[1,2-a] indol-1-one scaffold. Bioorg. Med. Chem. Lett. 2010, 20, 5713–5717.
- Abbiati, G.; Arcadi, A.; Beccalli, E.; Rossi, E. Novel intramolecular cyclization of N-alkynyl heterocycles containing proximate nucleophiles. Tetrahedron Lett. 2003, 44, 5331–5334.
- Abbiati, G.; Arcadi, A.; Bellinazzi, A.; Beccalli, E.; Rossi, E.; Zanzola, S. Intramolecular cyclization of δ-iminoacetylenes: A new entry to pyrazino[1,2-a] indoles. J. Org. Chem. 2005, 70, 4088–4095.
- Festa, A.A.; Zalte, R.R.; Golantsov, N.E.; Varlamov, A.V.; Van der Eycken, E.V.; Voskressensky, L.G. DBU-catalyzed alkyne—Imidate cyclization toward 1-alkoxypyrazino[1,2-a] indole synthesis. J. Org. Chem. 2018, 83, 9305–9311.
- Guven, S.; Ozer, M.S.; Kaya, S.; Menges, N.; Balci, M. Gold-catalyzed oxime–oxime rearrangement. Org. Lett. 2015, 17, 2660–2663.
- Bi, H.Y.; Du, M.; Pan, C.X.; Xiao, Y.; Su, G.F.; Mo, D.L. Nickel(II)-catalyzed [5 + 1] annulation of 2-carbonyl-1-propargylindoles with hydroxylamine to synthesize pyrazino[1,2-a] indole-2-oxides in water. J. Org. Chem. 2019, 84, 9859–9868.
- Basceken, S. Kaya, S.; Balci, M. Intramolecular gold-catalyzed and NaH-supported cyclization reactions of N-propargyl indole derivatives with pyrazole and pyrrole rings: Synthesis of pyrazolodiazepinoindole, pyrazolopyrazinoindole, and pyrrolopyrazinoindole. J. Org. Chem. 2015, 80, 12552–12561.
- Festa, A.A.; Golantsov, N.E.; Storozhenko, O.A.; Shumsky, A.N.; Varlamov, A.V.; Voskressensky, L.G. Alcohol-initiated dinitrile cyclization in basic media: A route toward pyrazino [1,2-a] indole-3-amines. Synlett 2018, 29, 898–903.
- Nayak, M.; Pandey, G.; Batra, S. Synthesis of pyrrolo[1,2-a] pyrazines and pyrazino[1,2-a] indoles by curtius reaction in morita–baylis–hillman derivatives. Tetrahedron 2011, 67, 7563–7569.
- Katritzky, A.R.; Verma, A.K.; He, H.Y.; Chandra, R. Novel synthesis of 1,2,3,4-tetrahydropyrazino[1,2-a] indoles. J. Org. Chem. 2003, 68, 4938–4940.
- Tiwari, R.K.; Singh, J.; Singh, D.; Verma, A.K.; Chandra, R. Highly efficient one-pot synthesis of 1-substituted-1,2,3,4-tetrahydropyrazino [1,2-a] indoles. Tetrahedron 2005, 61, 9513–9518.
- Salahi, S.; Ghandi, M.; Abbasi, A. An efficient ugi-azide four-component approach for the preparation of novel 1-(1H-tetrazol-5-yl)-10-chloro-1,2,3,4-tetrahydropyrazino[1,2-a] indoles. J. Heterocyclic Chem. 2019, 56, 1296–1305.
- Schönherr, H.; Leighton, J.L. Direct and highly enantioselective iso-pictet-spengler reactions with α-ketoamides: Access to underexplored indole core structures. Org. Lett. 2012, 14, 2610–2613.
- Milcendeau, P.; Zhang, Z.; Glinsky-Olivier, N.; van Elslande, E.; Guinchard, X. Au(I)-catalyzed pictet-spengler reactions all around the indole ring. J. Org. Chem. 2021, 86, 6406–6422.
- Guarnieri-Ibáňez, A.; de Aguirre, A.; Besnard, C.; Poblador-Bahamonde, A.I.; Lacour, J. Regiodivergent synthesis of pyrazino-indolines vs. triazocines via α-imino carbenes addition to imidazolidines. Chem. Sci. 2021, 12, 1479–1485.
- Wani, I.A.; Das, S.; Mondal, S.; Ghorai, M.K. Stereoselective construction of pyrazinoindoles and oxazinoindoles via ring-opening/pictet-spengler reaction of aziridines and epoxides with 3-methylindoles and carbonyls. J. Org. Chem. 2018, 83, 14553–14567.
- Singh, A.; Singh, S.; Sewariya, S.; Singh, N.; Singh, P.; Kumar, A.; Bandichhor, R.; Chandra, R. Stereospecific N-acylation of indoles and corresponding microwave mediated synthesis of pyrazinoindoles using hexafluoroisopropanol. Tetrahedron 2021, 84, 132017.
- Bos, M.; Jenck, F.; Martin, J.R.; Moreau, J.L.; Mutel, V.; Sleight, A.J.; Widmer, U. Synthesis, pharmacology and therapeutic potential of 10-methoxypyrazino[1,2-a] indoles, partial agonists at the 5HT2c receptor. Eur. J. Med. Chem. 1997, 32, 253–261.
- Chang-Fong, J.; Tyacke, R.J.; Lau, A.; Westaway, J.; Hudson, A.L.; Glennon, R.A. Pyrazino [1,2-a] indoles as novel high-affinity and selective imidazoline I2 receptor ligands. Bioorg. Med. Chem. Lett. 2004, 14, 1003–1005.
- Husbands, S.M.; Glennon, R.A.; Gorgerat, S.; Gough, R.; Tyacke, R.; Crosby, J.; Nutt, D.J.; Lewis, J.W.; Hudson, A.L. β-carboline binding to imidazoline receptors. Drug Alcohol Depend. 2001, 64, 203–208.
- Glennon, R.A.; Grella, B.; Tyacke, R.J.; Lau, A.; Westaway, J.; Hudson, A.L. Binding of β-carbolines at imidazoline I2 receptors: A structure–affinity investigation. Bioorg. Med. Chem. Lett. 2004, 14, 999–1002.
- Guandalini, L.; Martini, E.; Gualtieri, F.; Romanelli, M.N.; Varani, K. Design, synthesis and preliminary pharmacological evaluation of rigid analogues of the nicotinic agonist 1,1-dimethyl-4-phenylpiperazinium iodide (DMPP). Arkivoc 2004, 2004, 286–300.
- Buzard, D.J.; Schrader, T.O.; Zhu, X.; Lehmann, J.; Johnson, B.; Kasem, M.; Kim, S.H.; Kawasaki, A.; Lopez, L.; Moody, J.; et al. Design and synthesis of new tricyclic indoles as potent modulators of the S1P1 receptor. Bioorg. Med. Chem. Lett. 2015, 25, 659–663.
- Buzard, D.J.; Kim, S.H.; Lopez, L.; Kawasaki, A.; Zhu, X.; Moody, J.; Thoresen, T.; Calderon, I.; Ullman, B.; Han, S.; et al. Discovery of APD334: Design of a clinical stage functional antagonist of the sphingosine-1-phosphate-1 receptor. ACS Med. Chem. Lett. 2014, 5, 1313–1317.
- Bit, R.A.; Davis, P.D.; Elliott, L.H.; Harris, W.; Hill, C.H.; Keech, E.; Kumar, H.; Lawton, G.; Maw, A.; Nixon, J.S.; et al. Inhibitors of protein kinase C. 3. Potent and highly selective bisindolylmaleimides by conformational restriction. J. Med. Chem. 1993, 36, 21–29.
- Tiwari, R.K.; Singh, D.; Singh, J.; Yadav, V.; Pathak, A.K.; Dabur, R.; Chhillar, A.K.; Singh, R.; Sharma, G.L.; Chambra, R.; et al. Synthesis and antibacterial activity of substituted 1,2,3,4-tetrahydropyrazino [1,2-a] indoles. Bioorg. Med. Chem. Lett. 2006, 16, 413–416.
- Tiwari, R.K.; Verma, A.K.; Chhillar, A.K.; Singh, D.; Singh, J.; Sankar, V.K.; Yadav, V.; Sharma, G.L.; Chandra, R. Synthesis and antibacterial activity of substituted 1,2,3,4-tetrahydropyrazino[1,2-a] indoles. Bioorg. Med. Chem. 2006, 14, 2747–2752.
- Romagnoli, R.; Baraldi, P.G.; Carrion, M.D.; Cruz-Lopez, O.; Lopez Cara, C.; Preti, D.; Tabrizi, M.A. Balzarini, J.; Hamel, E.; Fabbri, E.; et al. Discovery of 8-methoxypyrazino [1,2-a] indole as a new potent antiproliferative agent against human leukemia K562 cells. A structure-activity relationship study. Lett. Drug Des. Discov. 2009, 6, 298–303.
- Kumar, K.S.; Kumar, N.P.; Rajesham, B.; Kishan, G.; Akula, S.; Kancha, R.K. Silver-catalyzed synthesis of pyrrolopiperazine fused with oxazine/imidazole via a domino approach: Evaluation of anti-cancer activity. N. J. Chem. 2018, 42, 34–38.
More
Information
Subjects:
Chemistry, Medicinal
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.2K
Entry Collection:
Organic Synthesis
Revisions:
2 times
(View History)
Update Date:
10 Aug 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No