 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Haruki Koike | + 2109 word(s) | 2109 | 2021-07-06 09:49:11 | | | |
| 2 | Vivi Li | + 130 word(s) | 2239 | 2021-07-19 05:01:35 | | |
Video Upload Options
Transthyretin (TTR) amyloidosis is caused by systemic deposition of wild-type or variant amyloidogenic TTR (ATTRwt and ATTRv, respectively). ATTRwt amyloidosis has traditionally been termed senile systemic amyloidosis, while ATTRv amyloidosis has been called familial amyloid polyneuropathy. Although ATTRwt amyloidosis has classically been regarded as one of the causes of cardiomyopathy occurring in the elderly population, recent developments in diagnostic techniques have significantly expanded the concept of this disease. For example, this disease is now considered an important cause of carpal tunnel syndrome in the elderly population. The phenotypes of ATTRv amyloidosis also vary depending on the mutation and age of onset. Peripheral neuropathy usually predominates in patients from the conventional endemic foci, while cardiomyopathy or oculoleptomeningeal involvement may also become major problems in other patients. Electron microscopic studies indicate that the direct impact of amyloid fibrils on surrounding tissues leads to organ damage, whereas accumulating evidence suggests that nonfibrillar TTR, such as oligomeric TTR, is toxic, inducing neurodegeneration. Microangiopathy has been suggested to act as an initial lesion, increasing the leakage of circulating TTR. Regarding treatments, the efficacy of liver transplantation has been established for ATTRv amyloidosis patients, particularly patients with early-onset amyloidosis. Recent phase III clinical trials have shown the efficacy of TTR stabilizers, such as tafamidis and diflunisal, for both ATTRwt and ATTRv amyloidosis patients.
1. Introduction
2. Diversity of Clinical Features
| Features | Early-Onset Patients from Endemic Foci | Late-Onset Patients from Nonendemic Areas |
|---|---|---|
| Age of onset | Late 20s to early 40s | ≥50 years |
| Sex | Male = female | Male > female |
| Family history | Common | Frequently absent |
| Penetrance rate | High | Low |
| Cardiac involvement | Conduction defects | Heart failure |
| Sensory dissociation | Common | Rare |
| Autonomic dysfunction | Severe | Mild |
| in early disease stage | ||
| Modality of nerve fiber loss | Small > large | Small = large |
| Amount of amyloid deposits | Large | Small |
| in the peripheral nervous system | ||
| Length of amyloid fibrils | Long | Short |
3. Characteristics of Amyloid Fibrils Determining the Clinicopathological Features
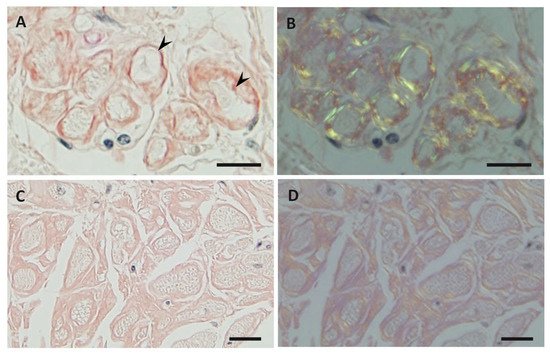
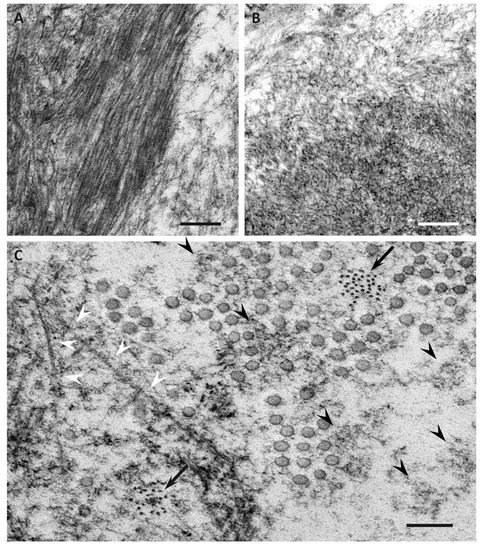
4. Impact of Amyloid Fibril Formation on Neighboring Tissues
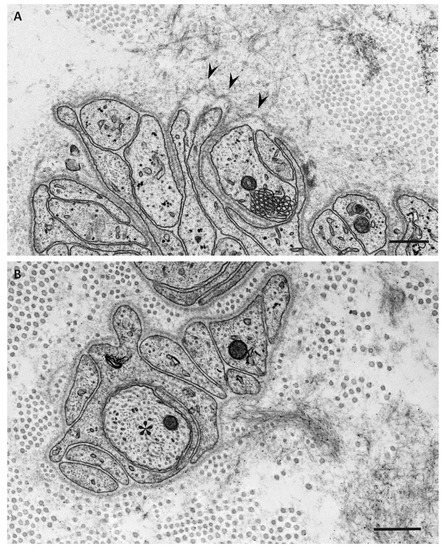
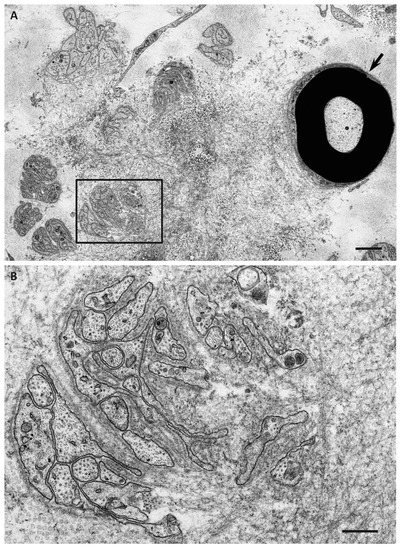
References
- Pitkänen, P.; Westermark, P.; Cornwell, G.G., 3rd. Senile systemic amyloidosis. Am. J. Pathol. 1984, 117, 391–399.
- Koike, H.; Misu, K.; Ikeda, S.; Ando, Y.; Nakazato, M.; Ando, E.; Yamamoto, M.; Hattori, N.; Sobue, G.; Study Group for Hereditary Neuropathy in Japan. Type I (transthyretin Met30) familial amyloid polyneuropathy in Japan: Early- vs. late-onset form. Arch. Neurol. 2002, 59, 1771–1776.
- Benson, M.D.; Kincaid, J.C. The molecular biology and clinical features of amyloid neuropathy. Muscle Nerve 2007, 36, 411–423.
- Planté-Bordeneuve, V.; Said, G. Familial amyloid polyneuropathy. Lancet Neurol. 2011, 10, 1086–1097.
- Adams, D.; Cauquil, C.; Labeyrie, C. Familial amyloid polyneuropathy. Curr. Opin. Neurol. 2017, 30, 481–489.
- Andrade, C. A peculiar form of peripheral neuropathy; familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain 1952, 75, 408–427.
- Araki, S.; Mawatari, S.; Ohta, M.; Nakajima, A.; Kuroiwa, Y. Polyneuritic amyloidosis in a Japanese family. Arch. Neurol. 1968, 18, 593–602.
- Andersson, R. Hereditary amyloidosis with polyneuropathy. Acta Med. Scand. 1970, 1–2, 85–94.
- Ando, Y.; Nakamura, M.; Araki, S. Transthyretin-related familial amyloidotic polyneuropathy. Arch. Neurol. 2005, 62, 1057–1062.
- Koike, H.; Tanaka, F.; Hashimoto, R.; Tomita, M.; Kawagashira, Y.; Iijima, M.; Fujitake, J.; Kawanami, T.; Kato, T.; Yamamoto, M.; et al. Natural history of transthyretin Val30Met familial amyloid polyneuropathy: Analysis of late-onset cases from non-endemic areas. J. Neurol. Neurosurg. Psychiatry 2012, 83, 152–158.
- Parman, Y.; Adams, D.; Obici, L.; Galán, L.; Guergueltcheva, V.; Suhr, O.B.; Coelho, T. European Network for TTR-FAP (ATTReuNET). Sixty years of transthyretin familial amyloid polyneuropathy (TTR-FAP) in Europe: Where are we now? A European network approach to defining the epidemiology and management patterns for TTR-FAP. Curr. Opin. Neurol. 2016, 29 (Suppl. 1), S3–S13.
- Sekijima, Y.; Ueda, M.; Koike, H.; Misawa, S.; Ishii, T.; Ando, Y. Diagnosis and management of transthyretin familial amyloid polyneuropathy in Japan: Red-flag symptom clusters and treatment algorithm. Orphanet J. Rare Dis. 2018, 13, 6.
- Chao, C.C.; Huang, C.M.; Chiang, H.H.; Luo, K.R.; Kan, H.W.; Yang, N.C.; Chiang, H.; Lin, W.M.; Lai, S.M.; Lee, M.J.; et al. Sudomotor innervation in transthyretin amyloid neuropathy: Pathology and functional correlates. Ann. Neurol. 2015, 78, 272–283.
- Carr, A.S.; Pelayo-Negro, A.L.; Evans, M.R.; Laurà, M.; Blake, J.; Stancanelli, C.; Iodice, V.; Wechalekar, A.D.; Whelan, C.J.; Gillmore, J.D.; et al. A study of the neuropathy associated with transthyretin amyloidosis (ATTR) in the UK. J. Neurol. Neurosurg. Psychiatry 2016, 87, 620–627.
- Durmuş-Tekçe, H.; Matur, Z.; Mert Atmaca, M.; Poda, M.; Çakar, A.; Hıdır Ulaş, Ü.; Oflazer-Serdaroğlu, P.; Deymeer, F.; Parman, Y.G. Genotypic and phenotypic presentation of transthyretin-related familial amyloid polyneuropathy (TTR-FAP) in Turkey. Neuromuscul. Disord. 2016, 26, 441–446.
- Holmgren, G.; Steen, L.; Ekstedt, J.; Groth, C.G.; Ericzon, B.G.; Eriksson, S.; Andersen, O.; Karlberg, I.; Nordén, G.; Nakazato, M.; et al. Biochemical effect of liver transplantation in two Swedish patients with familial amyloidotic polyneuropathy (FAP-met30). Clin. Genet. 1991, 40, 242–246.
- Yamashita, T.; Ando, Y.; Okamoto, S.; Misumi, Y.; Hirahara, T.; Ueda, M.; Obayashi, K.; Nakamura, M.; Jono, H.; Shono, M.; et al. Long-term survival after liver transplantation in patients with familial amyloid polyneuropathy. Neurology 2012, 78, 637–643.
- Coelho, T.; Maia, L.F.; Martins da Silva, A.; Waddington Cruz, M.; Planté-Bordeneuve, V.; Lozeron, P.; Suhr, O.B.; Campistol, J.M.; Conceição, I.M.; Schmidt, H.H.; et al. Tafamidis for transthyretin familial amyloid polyneuropathy: A randomized, controlled trial. Neurology 2012, 79, 785–792.
- Berk, J.L.; Suhr, O.B.; Obici, L.; Sekijima, Y.; Zeldenrust, S.R.; Yamashita, T.; Heneghan, M.A.; Gorevic, P.D.; Litchy, W.J.; Wiesman, J.F.; et al. Repurposing diflunisal for familial amyloid polyneuropathy: A randomized clinical trial. JAMA 2013, 310, 2658–2667.
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.M.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N. Engl. J. Med. 2018, 379, 1007–1016.
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018, 379, 11–21.
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Planté-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018, 379, 22–31.
- Koike, H.; Misu, K.; Sugiura, M.; Iijima, M.; Mori, K.; Yamamoto, M.; Hattori, N.; Mukai, E.; Ando, Y.; Ikeda, S.; et al. Pathology of early- vs. late-onset TTR Met30 familial amyloid polyneuropathy. Neurology 2004, 63, 129–138.
- Cornwell, G.G., 3rd; Murdoch, W.L.; Kyle, R.A.; Westermark, P.; Pitkänen, P. Frequency and distribution of senile cardiovascular amyloid. A clinicopathologic correlation. Am. J. Med. 1983, 75, 618–623.
- Tanskanen, M.; Peuralinna, T.; Polvikoski, T.; Notkola, I.L.; Sulkava, R.; Hardy, J.; Singleton, A.; Kiuru-Enari, S.; Paetau, A.; Tienari, P.J.; et al. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in α2-macroglobulin and tau: A population-based autopsy study. Ann. Med. 2008, 40, 232–239.
- Ueda, M.; Horibata, Y.; Shono, M.; Misumi, Y.; Oshima, T.; Su, Y.; Tasaki, M.; Shinriki, S.; Kawahara, S.; Jono, H.; et al. Clinicopathological features of senile systemic amyloidosis: An ante- and post-mortem study. Mod. Pathol. 2011, 24, 1533–1544.
- Sekijima, Y.; Yazaki, M.; Ueda, M.; Koike, H.; Yamada, M.; Ando, Y. First nationwide survey on systemic wild-type ATTR amyloidosis in Japan. Amyloid 2018, 25, 8–10.
- Grogan, M.; Scott, C.G.; Kyle, R.A.; Zeldenrust, S.R.; Gertz, M.A.; Lin, G.; Klarich, K.W.; Miller, W.L.; Maleszewski, J.J.; Dispenzieri, A. Natural history of wild-type transthyretin cardiac amyloidosis and risk stratification using a novel staging system. J. Am. Coll. Cardiol. 2016, 68, 1014–1020.
- Westermark, P.; Westermark, G.T.; Suhr, O.B.; Berg, S. Transthyretin-derived amyloidosis: Probably a common cause of lumbar spinal stenosis. Ups J. Med. Sci. 2014, 119, 223–228.
- Yanagisawa, A.; Ueda, M.; Sueyoshi, T.; Okada, T.; Fujimoto, T.; Ogi, Y.; Kitagawa, K.; Tasaki, M.; Misumi, Y.; Oshima, T.; et al. Amyloid deposits derived from transthyretin in the ligamentum flavum as related to lumbar spinal canal stenosis. Mod. Pathol. 2015, 28, 201–207.
- Koike, H.; Kawagashira, Y.; Iijima, M.; Yamamoto, M.; Hattori, N.; Tanaka, F.; Hirayama, M.; Ando, Y.; Ikeda, S.; Sobue, G. Electrophysiological features of late-onset transthyretin Met30 familial amyloid polyneuropathy unrelated to endemic foci. J. Neurol. 2008, 255, 1526–1533.
- Koike, H.; Morozumi, S.; Kawagashira, Y.; Iijima, M.; Yamamoto, M.; Hattori, N.; Tanaka, F.; Nakamura, T.; Hirayama, M.; Ando, Y.; et al. The significance of carpal tunnel syndrome in transthyretin Val30Met familial amyloid polyneuropathy. Amyloid 2009, 16, 142–148.
- Yamashita, T.; Ueda, M.; Misumi, Y.; Masuda, T.; Nomura, T.; Tasaki, M.; Takamatsu, K.; Sasada, K.; Obayashi, K.; Matsui, H.; et al. Genetic and clinical characteristics of hereditary transthyretin amyloidosis in endemic and non-endemic areas: Experience from a single-referral center in Japan. J. Neurol. 2018, 265, 134–140.
- Koike, H.; Sobue, G. Diagnosis of familial amyloid polyneuropathy: Wide-ranged clinicopathological features. Expert Opin. Med. Diagn. 2010, 4, 323–331.
- Koike, H.; Sobue, G. Late-onset familial amyloid polyneuropathy in Japan. Amyloid 2012, 19 (Suppl. 1), 55–57.
- Lemos, C.; Coelho, T.; Alves-Ferreira, M.; Martins-da-Silva, A.; Sequeiros, J.; Mendonça, D.; Sousa, A. Overcoming artefact: Anticipation in 284 Portuguese kindreds with familial amyloid polyneuropathy (FAP) ATTRV30M. J. Neurol. Neurosurg. Psychiatry 2014, 85, 326–330.
- Koike, H.; Hashimoto, R.; Tomita, M.; Kawagashira, Y.; Iijima, M.; Tanaka, F.; Sobue, G. Diagnosis of sporadic transthyretin Val30Met familial amyloid polyneuropathy: A practical analysis. Amyloid 2011, 18, 53–62.
- Misu, K.; Hattori, N.; Nagamatsu, M.; Ikeda, S.; Ando, Y.; Nakazato, M.; Takei, Y.; Hanyu, N.; Usui, Y.; Tanaka, F.; et al. Late-onset familial amyloid polyneuropathy type I (transthyretin Met30-associated familial amyloid polyneuropathy) unrelated to endemic focus in Japan. Clinicopathological and genetic features. Brain 1999, 122, 1951–1962.
- Misu, K.; Hattori, N.; Ando, Y.; Ikeda, S.; Sobue, G. Anticipation in early- but not late-onset familial amyloid polyneuropathy (TTR met 30) in Japan. Neurology 2000, 55, 451–452.
- Koike, H.; Ikeda, S.; Takahashi, M.; Kawagashira, Y.; Iijima, M.; Misumi, Y.; Ando, Y.; Ikeda, S.I.; Katsuno, M.; Sobue, G. Schwann cell and endothelial cell damage in transthyretin familial amyloid polyneuropathy. Neurology 2016, 87, 2220–2229.
- Ihse, E.; Ybo, A.; Suhr, O.; Lindqvist, P.; Backman, C.; Westermark, P. Amyloid fibril composition is related to the phenotype of hereditary transthyretin V30M amyloidosis. J. Pathol. 2008, 216, 253–261.
- Ihse, E.; Rapezzi, C.; Merlini, G.; Benson, M.D.; Ando, Y.; Suhr, O.B.; Ikeda, S.; Lavatelli, F.; Obici, L.; Quarta, C.C.; et al. Amyloid fibrils containing fragmented ATTR may be the standard fibril composition in ATTR amyloidosis. Amyloid 2013, 20, 142–150.
- Koike, H.; Ando, Y.; Ueda, M.; Kawagashira, Y.; Iijima, M.; Fujitake, J.; Hayashi, M.; Yamamoto, M.; Mukai, E.; Nakamura, T.; et al. Distinct characteristics of amyloid deposits in early- and late-onset transthyretin Val30Met familial amyloid polyneuropathy. J. Neurol. Sci. 2009, 287, 178–184.
- Koike, H.; Nishi, R.; Ikeda, S.; Kawagashira, Y.; Iijima, M.; Sakurai, T.; Shimohata, T.; Katsuno, M.; Sobue, G. The morphology of amyloid fibrils and their impact on tissue damage in hereditary transthyretin amyloidosis: An ultrastructural study. J. Neurol. Sci. 2018, 394, 99–106.
- Bergström, J.; Gustavsson, A.; Hellman, U.; Sletten, K.; Murphy, C.L.; Weiss, D.T.; Solomon, A.; Olofsson, B.O.; Westermark, P. Amyloid deposits in transthyretin-derived amyloidosis: Cleaved transthyretin is associated with distinct amyloid morphology. J. Pathol. 2005, 206, 224–232.
- Yazaki, M.; Mitsuhashi, S.; Tokuda, T.; Kametani, F.; Takei, Y.I.; Koyama, J.; Kawamorita, A.; Kanno, H.; Ikeda, S.I. Progressive wild-type transthyretin deposition after liver transplantation preferentially occurs onto myocardium in FAP patients. Am. J. Transplant. 2007, 7, 235–242.
- Okamoto, S.; Wixner, J.; Obayashi, K.; Ando, Y.; Ericzon, B.G.; Friman, S.; Uchino, M.; Suhr, O.B. Liver transplantation for familial amyloidotic polyneuropathy: Impact on Swedish patients’ survival. Liver Transplant. 2009, 15, 1229–1235.
- Suhr, O.B.; Lundgren, E.; Westermark, P. One mutation, two distinct disease variants: Unravelling the impact of transthyretin amyloid fibril composition. J. Intern. Med. 2017, 281, 337–347.
- Mangione, P.P.; Verona, G.; Corazza, A.; Marcoux, J.; Canetti, D.; Giorgetti, S.; Raimondi, S.; Stoppini, M.; Esposito, M.; Relini, A.; et al. Plasminogen activation triggers transthyretin amyloidogenesis in vitro. J. Biol. Chem. 2018, 293, 14192–14199.
- Oshima, T.; Kawahara, S.; Ueda, M.; Kawakami, Y.; Tanaka, R.; Okazaki, T.; Misumi, Y.; Obayashi, K.; Yamashita, T.; Ohya, Y.; et al. Changes in pathological and biochemical findings of systemic tissue sites in familial amyloid polyneuropathy more than 10 years after liver transplantation. J. Neurol. Neurosurg. Psychiatry 2014, 85, 740–746.
- Marcoux, J.; Mangione, P.P.; Porcari, R.; Degiacomi, M.T.; Verona, G.; Taylor, G.W.; Giorgetti, S.; Raimondi, S.; Sanglier-Cianférani, S.; Benesch, J.L.; et al. A novel mechano-enzymatic cleavage mechanism underlies transthyretin amyloidogenesis. EMBO Mol. Med. 2015, 7, 1337–1349.
- Sueyoshi, T.; Ueda, M.; Jono, H.; Irie, H.; Sei, A.; Ide, J.; Ando, Y.; Mizuta, H. Wild-type transthyretin-derived amyloidosis in various ligaments and tendons. Hum. Pathol. 2011, 42, 1259–1264.
- Misumi, Y.; Ando, Y.; Ueda, M.; Obayashi, K.; Jono, H.; Su, Y.; Yamashita, T.; Uchino, M. Chain reaction of amyloid fibril formation with induction of basement membrane in familial amyloidotic polyneuropathy. J. Pathol. 2009, 219, 481–490.
- Hou, X.; Richardson, S.J.; Aguilar, M.I.; Small, D.H. Binding of amyloidogenic transthyretin to the plasma membrane alters membrane fluidity and induces neurotoxicity. Biochemistry 2005, 44, 11618–11627.

