Transthyretin (TTR) amyloidosis is caused by systemic deposition of wild-type or variant amyloidogenic TTR (ATTRwt and ATTRv, respectively). ATTRwt amyloidosis has traditionally been termed senile systemic amyloidosis, while ATTRv amyloidosis has been called familial amyloid polyneuropathy. Although ATTRwt amyloidosis has classically been regarded as one of the causes of cardiomyopathy occurring in the elderly population, recent developments in diagnostic techniques have significantly expanded the concept of this disease. For example, this disease is now considered an important cause of carpal tunnel syndrome in the elderly population. The phenotypes of ATTRv amyloidosis also vary depending on the mutation and age of onset. Peripheral neuropathy usually predominates in patients from the conventional endemic foci, while cardiomyopathy or oculoleptomeningeal involvement may also become major problems in other patients. Electron microscopic studies indicate that the direct impact of amyloid fibrils on surrounding tissues leads to organ damage, whereas accumulating evidence suggests that nonfibrillar TTR, such as oligomeric TTR, is toxic, inducing neurodegeneration. Microangiopathy has been suggested to act as an initial lesion, increasing the leakage of circulating TTR. Regarding treatments, the efficacy of liver transplantation has been established for ATTRv amyloidosis patients, particularly patients with early-onset amyloidosis. Recent phase III clinical trials have shown the efficacy of TTR stabilizers, such as tafamidis and diflunisal, for both ATTRwt and ATTRv amyloidosis patients.
- angiopathy
- diflunisal
- electron microscopy
- oligomers
- pathogenesis
- pathology
- protein misfolding disease
- Schwann cell
- tafamidis
- therapy
1. Introduction
2. Diversity of Clinical Features
As ATTR amyloidosis is a systemic disease, patients exhibit variable clinical features depending on the site of amyloid deposition [23][34]. ATTRwt amyloidosis has classically been regarded as one of the causes of cardiomyopathy in the elderly population. Studies of autopsy specimens revealed that a significant proportion of the elderly population have wild-type TTR deposition, particularly in the heart (12 to 25% of subjects aged >80 years), despite a lack of relevant symptoms [24][25][26][35,36,37]. However, the recent development of diagnostic techniques for amyloidosis has significantly expanded the concept of this disease [27][38]. For example, this disease is now considered an important cause of carpal tunnel syndrome in the elderly population [27][28][38,39]. Some studies have also suggested an association between wild-type TTR deposition in ligaments and spinal canal stenosis [27][29][30][38,40,41]. The phenotypes of ATTRv amyloidosis are also variable, depending on the mutation and age at onset [2][12][2,12]. As the classical name “familial amyloid polyneuropathy” indicates, peripheral neuropathy usually predominates in patients with conventional endemic foci [31][32][42,43]. Cardiomyopathy or oculoleptomeningeal involvement may also become major problems in others, particularly in patients with non-Val30Met mutations [12][33][12,44]. For example, Val112Ile and Thr60Ala mutations are usually associated with cardiac amyloidosis, while Tyr114Cys mutation causes oculoleptomeningeal amyloidosis [12]. Regarding the most common mutation, Val30Met (i.e., ATTR Val30Met amyloidosis), patients from the conventional endemic foci of Portugal and Japan exhibit textbook features of amyloid neuropathy, such as the following: early disease onset ranging in age from the late 20s to early 40s; a high penetrance rate; a nearly 1-to-1 male-to-female ratio; marked autonomic dysfunction; loss of superficial sensation, including nociception and thermal sensation (i.e., sensory dissociation); atrioventricular conduction block requiring pacemaker implantation; and the presence of anticipation of age at onset (Table 1) [2][34][35][36][2,45,46,47]. By contrast, patients with Val30Met mutations from nonendemic areas exhibit an older age at disease onset of over 50 years, a low penetrance rate, extreme male preponderance, relatively mild autonomic dysfunction, loss of all sensory modalities rather than sensory dissociation, the frequent presence of cardiomegaly, and the absence of anticipation of age at onset [2][10][37][38][39][2,10,48,49,50]. Despite the presence of the same mutation in the TTR gene, the reason for the differential clinical features between early- and late-onset cases has not been clarified.| Features | Early-Onset Patients from Endemic Foci | Late-Onset Patients from Nonendemic Areas |
|---|---|---|
| Age of onset | Late 20s to early 40s | ≥50 years |
| Sex | Male = female | Male > female |
| Family history | Common | Frequently absent |
| Penetrance rate | High | Low |
| Cardiac involvement | Conduction defects | Heart failure |
| Sensory dissociation | Common | Rare |
| Autonomic dysfunction | Severe | Mild |
| in early disease stage | ||
| Modality of nerve fiber loss | Small > large | Small = large |
| Amount of amyloid deposits | Large | Small |
| in the peripheral nervous system | ||
| Length of amyloid fibrils | Long | Short |
3. Characteristics of Amyloid Fibrils Determining the Clinicopathological Features
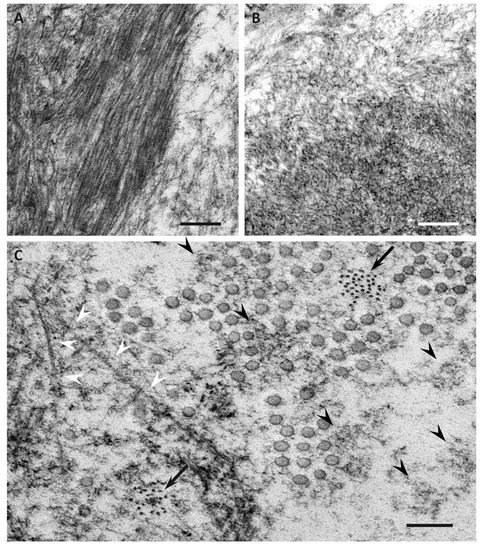
Previous studies have demonstrated differences in the characteristics of amyloid fibrils depending on the age of onset and the type of mutation in patients with ATTRv amyloidosis [40][41][42][43][44][51,53,54,55,56]. In early-onset Val30Met cases, long and thick amyloid fibrils are common (Figure 2A), whereas the fibrils are usually short and thin in late-onset Val30Met cases and most non-Val30Met cases (Figure 2B) [40][42][44][51,54,56]. In addition, amyloid deposits in early-onset Val30Met cases tend to be highly congophilic and show strong apple-green birefringence, while those in late-onset Val30Met cases are generally weakly congophilic and show faint apple-green birefringence (Figure 1) [43][55]. These differences in the characteristics of amyloid deposits between early- and late-onset cases are particularly conspicuous in the heart [41][43][53,55]. Interestingly, short amyloid fibrils and a weak affinity of amyloid deposits for Congo red have also been reported for cardiac amyloid deposits in patients with ATTRwt amyloidosis [45][57]. A study of autopsied Japanese Val30Met patients demonstrated that most TTR in cardiac amyloid deposits from the early-onset cases was variant TTR, whereas wild-type TTR constituted more than half of the TTR in the deposits from the late-onset cases [43][55]. In ATTRv amyloidosis patients who undergo liver transplantation, cardiac amyloidosis may progress even after transplantation due to wild-type TTR deposition, particularly in elderly male patients [46][47][58,59]. These findings suggest that the mechanism of amyloid deposition in the heart is similar between late-onset ATTRv amyloidosis patients and ATTRwt amyloidosis patients. Interestingly, ATTRwt amyloidosis mainly affects males, who account for approximately 90% of patients [27][28][38,39]. This male preponderance is in accordance with late-onset ATTR Val30Met amyloidosis cases [10], but not with early-onset Val30Met cases, which show a nearly 1-to-1 male-to-female ratio [31][42].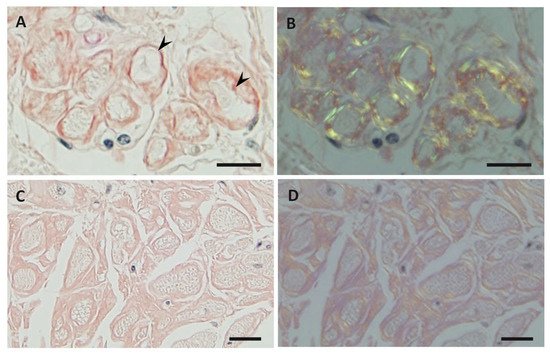
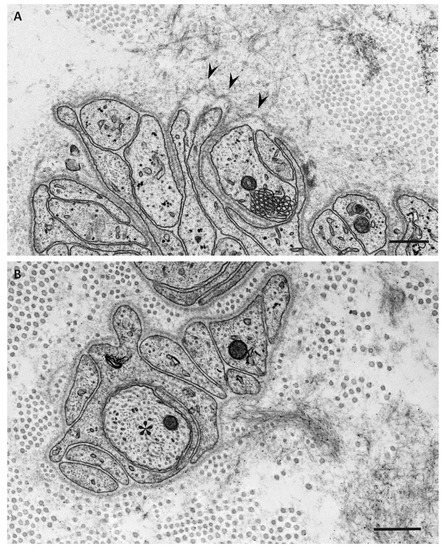
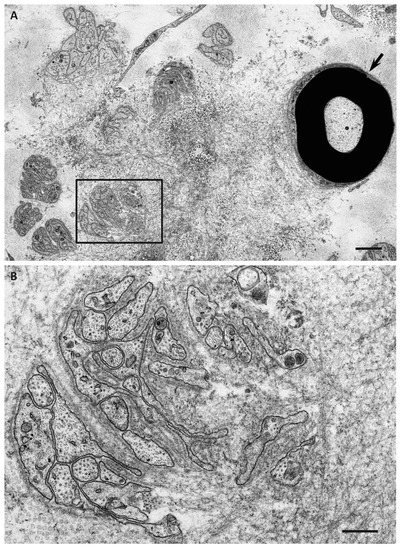
4. Impact of Amyloid Fibril Formation on Neighboring Tissues
Electron microscopic studies of nerve biopsy specimens from patients with ATTRv amyloidosis have shown that amyloid fibrils were formed among amorphous electron-dense materials located in extracellular spaces of the endoneurium [44][56]. Amorphous electron-dense materials tend to be observed around microvessels and the subperineurial space. Among these amorphous materials, dotty or fine fibrillar structures are frequently observed (Figure 2C). The dotty structures seem to be the core of amyloid fibrils because slightly elongated fibrillar structures with a thickness similar to the diameter of these dots are frequently found [44][56]. The mature long fibers usually occupy the central part of the large aggregations of amyloid fibrils, while the amorphous materials, dotty structures, and short amyloid fibrils tend to be present at the periphery of the aggregates of amyloid fibrils. During the process of amyloid fibril maturation, amyloid fibrils seem to pull surrounding tissues [44][56]. This traction of neighboring tissues seems to be conspicuous in cases with long and thick amyloid fibrils, such as early-onset Val30Met cases in endemic foci (Figure 3A) [40][44][51,56]. By contrast, amyloid fibril maturation seems to have a smaller influence on neighboring tissues in cases with short and fine amyloid fibrils, such as late-onset Val30Met cases in nonendemic areas (Figure 3B) [40][44][51,56].