 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Viet-Khoa Tran-Nguyen | + 5435 word(s) | 5435 | 2020-06-22 05:26:25 | | | |
| 2 | Viet-Khoa Tran-Nguyen | Meta information modification | 5435 | 2020-06-29 13:47:29 | | | | |
| 3 | Rita Xu | -899 word(s) | 4536 | 2020-07-16 11:08:52 | | | | |
| 4 | Rita Xu | -94 word(s) | 4442 | 2020-10-26 10:14:11 | | |
Video Upload Options
Developing realistic data sets for evaluating virtual screening methods is a task that has been tackled by the cheminformatics community for many years. Numerous artificially constructed data collections were developed, such as DUD, DUD-E, or DEKOIS. However, they all suffer from multiple drawbacks, one of which is the absence of experimental results confirming the impotence of presumably inactive molecules, leading to possible false negatives in the ligand sets. In light of this problem, the PubChem BioAssay database, an open-access repository providing the bioactivity information of compounds that were already tested on a biological target, is now a recommended source for data set construction. Nevertheless, there exist several issues with the use of such data that need to be properly addressed. In this article, an overview of benchmarking data collections built upon experimental PubChem BioAssay input is provided, along with a thorough discussion of noteworthy issues that one must consider during the design of new ligand sets from this database. The points raised in this review are expected to guide future developments in this regard, in hopes of offering better evaluation tools for novel in silico screening procedures.
1. Introduction
As demonstrated in the literature and the previous section, data retrieved from PubChem BioAssay may be used for various purposes in cheminformatics-related research, including benchmarking data set construction. Due to the availability of a wide range of assays with diverse ligand sets that the database offers, it is important to be conscious of all the issues that may arise regarding the usage of such large data [1][2][3], in terms of assay selection and data curation, to properly employ these abundant resources.
2. Assay Selection for Evaluating Virtual Screening Methods
2.1. Assay Selection as Regards the Data Size and Hit Rates
One of the first questions that we have to face when using data from the PubChem BioAssay repository to build benchmarking data sets concerns the assay(s) that should be chosen. As mentioned earlier in the manuscript, as of 30 April 2020, there were over a million assays deposited on the database. However, only a few of them can be deemed suitable for method evaluation purposes. There are many factors that one should consider before deciding which assay(s) to use. We herewith propose, as primary conditions to filter out unsuitable assays, the selection of only small-molecule HTS assays yielding biologically active molecules. RNAi assays, on the other hand, were conducted on microRNA-like molecules comprising twenties of base pairs that violate most drug-likeness rules of thumb and are, therefore, not of great interest in small-molecule drug discovery. For the sake of having an acceptable amount of ligands in the data that may give a meaningful retrospective evaluation of in silico screening methods, we recommend that only assays with no fewer than 10 actives selected among at least 300 tested substances should be kept. Data sets including only nine or fewer actives are considered too small and would be over-challenging for virtual screening, especially for machine-learning algorithms to learn anything meaningful. On the other hand, assays conducted with fewer than 300 substances while yielding more than 10 actives give hit rates that are deemed too high in comparison to those typically observed in experimental screening decks [1], even higher than those of existing data sets such as DUD [4], DUD-E [5], or DEKOIS 2.0 [6]. There may exist, of course, assays with high hit rates that remain after this initial check (e.g., AIDs 1, 3, 720690 and 720697); however, the aforementioned conditions are proposed to demonstrate that there is only a very small portion of available PubChem assays (0.20%) whose data may be considered for evaluating virtual screening protocols (Figure 1). The ligand sets of the remaining assays need to be further examined and may be filtered to ensure that their hit rates are as close as possible to those of experimental HTS campaigns and that they are suitable for the nature of the screening method (ligand-based or structure-based).
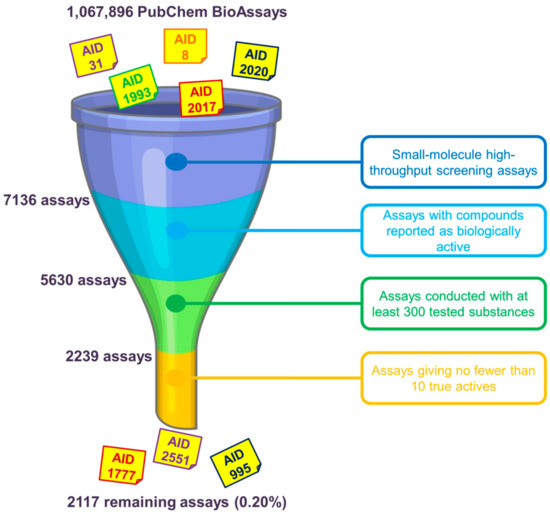
Figure 1. Primary selection of PubChem assays whose ligand sets should be further considered for evaluating virtual screening methods. We herewith recommend the use of only small-molecule high-throughput screening (HTS) assays giving at least 10 biologically active molecules among no fewer than 300 tested substances. Overall, there are only 2117 assays (0.20% of 1,067,896 assays in total, as of 30 April 2020) that remain, indicating a very small portion of PubChem assays that may be considered after this initial check.
2.2. Assay Selection as Regards the Nature of Virtual Screening
As demonstrated in various papers, a ligand set may be appropriate for the evaluation of only ligand-based in silico approaches [7][8], or only structure-based methods [9], or sometimes both [1][2][3]. This depends on the quantity and the chemical composition of all molecules that constitute the data set and the availability and the quality of 3-dimensional structures of relevant protein targets, as well as the definition of binding site(s) in which active substances exert their bioactivity. Data sets retrieved from the PubChem BioAssay database, being no exception, have to be thoroughly examined according to the criteria mentioned above before being used to assess a certain virtual screening method. Ideally speaking, an assay whose ligands are considered for evaluating structure-based approaches needs to be conducted on a protein target whose structure has been solved at a high resolution, with no ambiguity in terms of electron density, with at least a molecule of the same phenotype (agonist, antagonist, inhibitor, etc.) as that of the active compounds. However, targets for which no crystallographic or electron-microscopic structure is deposited on the Protein Data Bank may also be considered if high-quality homology models are available. An example of this can be seen in the assay AID 588606, featuring inhibitors of the yeast efflux pump Cdr1. Though the protein target, the ABC (ATP-binding cassette) drug-resistance protein 1 of Candida albicans (CaCdr1p), has not yet been available in the Protein Data Bank with a known inhibitor, a homology model of this transporter was generated using the human ABCG5/G8 crystal structure as the template, and possible binding sites located in the transmembrane domain were identified and validated by means of atomic modeling and systematic mutagenesis, confirming their essential role in Cdr1p-induced multidrug resistance [10]. However, caution should be taken when one uses such artificially constructed models as the input for structure-based screening approaches. On the other hand, the presence of many nonoverlapping binding sites (orthosteric versus allosteric) in the 3D structures of protein targets (as observed in those of AIDs 1469, 624170, or 624417), either crystallographic or not, may ultimately become a reason for failures in screening PubChem molecules on such proteins, especially when there is no information on the exact binding site of the tested substances that can be deduced from the assay description [1]. As virtual screening performances may vary quite significantly depending on the protein structure employed as the input [1], one should therefore be cautious when using data of these assays for evaluating structure-based screening procedures, lest they give poorer performances than expected due to external reasons that are not related to the methods themselves. Another point that should not be overlooked concerns assays that were conducted on substances derived from only a few chemical series, as they may give rise to bias that overestimates the screening performance, notably that of ligand-based approaches. If another similar assay on the same target but with a more diverse ligand set (in terms of chemical features) is available, one is recommended to make use of this assay instead. Otherwise, the “biased” data need further tuning to be deemed suitable for evaluation purposes, e.g., by filtering out “redundant” compounds (this point will be thoroughly discussed in the next section of this manuscript). However, this ligand-filtering process should not lower the number of active substances to a value so small that ligand-based methods or machine-learning algorithms cannot come up with meaningful results.
2.3. Assay Selection as Regards the Screening Stage
Additionally, the use of data from “primary assays” should be subject to caution, as the activity outcome was only determined at a single concentration and has not yet been validated on the basis of a dose-response relationship with multiple tested concentrations [11][12]; hence, the potency values of active molecules are not confirmed. As a matter of fact, some substances originally deemed as active in a primary assay may be denounced as inactive by a subsequent confirmatory screen, as seen in AIDs 449 and 466 or AIDs 524 and 548. We therefore recommend that primary screening data should only be used if there exists a confirmatory assay that validates the potency of the selected active molecules. This practice was already observed in the construction of the MUV data sets by Rohrer and Baumann [2], in which pairs of primary and corresponding confirmatory screens were employed, whose data were then combined to form the final ligand sets. In this manner, the large pool of inactive substances from the primary assay is not neglected, and the bioactivities of the confirmed hits are indeed guaranteed, affording a vast data set (usually implying a low hit rate) with fully validated active components. Otherwise, the output data of the primary screens alone should be used with great caution, due to the risk of assuming “false positives” that may later falsify the virtual screening outcomes. An exhaustive search on the whole PubChem BioAssay database is therefore of paramount importance to select relevant data sets for the retrospective assessment of in silico screening protocols in order to ensure the quality of such evaluations.
3. Detecting False Positives among Active Substances
Concerns have long been raised over the presence of chemical-induced artifacts in screening experiments, leading to false positive findings among the molecules deemed as active [1][2][3][13][14][15][16][17][18][19]. Misinterpretation of the assay results and subsequent inaccurate conclusions may stem from various reasons largely discussed in the literature. Among them are off-target effects of compounds exerting unspecific bioactivities, possible biological target precipitation by organic chemical aggregations, inherent fluorescent properties of substances that interfere with fluorescence emission detection methods, or luciferase inhibitory activities of molecules that spoil light emission measurements in reporter gene assays [2]. Active substances whose modes of action are subject to the aforementioned issues must therefore be removed from the PubChem BioAssay ligand sets before the data can be used for retrospective virtual screening purposes. Rohrer and Baumann (2009) addressed this problem during the construction of their MUV data sets from the database, designing a so-called “assay artifacts filter” aiming to eliminate all active ligands that likely become false positives, thus prevent them from affecting subsequent screening performances. The filter is composed of three filtering “layers”, including (i) the “Hill slope filter” after which the actives whose Hill slopes for the dose-response curves are lower than 0.5 or higher than 2 are eliminated, (ii) the “frequency of hits filter” that keeps only the molecules deemed as active in no more than 26% of the bioactivity assays in which they were tested, and (iii) the “auto-fluorescence and luciferase inhibition filter” that rules out compounds exhibiting auto-fluorescent properties along with inhibitors of luciferase [2]. All frequent hitters, unspecific binders (molecules with multiple binding sites), experimentally determined aggregators, and spoilers of optical detection methods are, as a result, removed from the PubChem data sets after these filtering steps. Such filters indeed have a profound impact on the population of active substances, as over a half of them were deleted by these “false positives filters” during the development of our recently introduced LIT-PCBA data set (Figure 4) [1]. This drastic decrease in the number of confirmed actives also helps lower the “hit rates” observed in our ligand sets (as only the actives were subjected to these filters), thus bringing them closer to those typically reported in high-throughput screening decks in reality and lower than those of artificially constructed data sets such as DUD [4], DUD-E [5], or DEKOIS 2.0 [6]. This not only denotes the particular challenge brought about by our data set but, also, highlights the importance of detecting and removing false positives in assembling active substances.
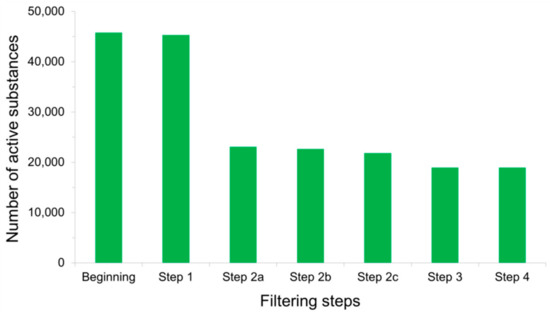
Figure 4. Total number of active substances that remained after each filtering step was applied to PubChem BioAssay ligands during the construction of the LIT-PCBA data set [1]: Step 1—inorganic molecules; Step 2a—actives with Hill slopes <0.5 or >2; Step 2b—actives with a frequency of hits >0.26; Step 2c—actives found among 10,892 confirmed aggregators, luciferase inhibitors, or auto-fluorescent molecules; Step 3—substances with extreme molecular properties; and Step 4—3D conversion and ionization failures. It can be observed that the sole step 2a removed the most active molecules (over 50% of them), thus significantly reducing the population of true actives in comparison to that of true inactives.
4. Possible Chemical Bias in Assembling Active and Inactive Substances
As previously mentioned, a noteworthy issue of raw data published on PubChem BioAssay lies in the chemically biased composition of active and inactive substances for a particular target. More specifically, there may exist “analog bias” [20] present among the molecules constituting a ligand set, which likely leads to overly good performances of virtual screening methods. This bias is generally observed in data collections whose actives (or inactives) share similar chemical features, meaning a large number of these molecules are issued from the same (or similar) scaffolds [9]. As ligand-based and structure-based screening methods tend to recognize compounds of the same chemical series, such bias may result in an overestimation of in silico screening performances [9]. Besides, significant differences between active and inactive molecules, in terms of physicochemical properties, such as molecular mass, octanol-water partition coefficient, or atomic formal charge, may as well be the source of artificial enrichment [2]. Raw experimental data from PubChem BioAssay therefore need to be finely tuned before further use, by filtering out most compounds representing the same scaffold while ensuring that the physicochemical parameters of all included molecules are kept within the same range, so that the chemical bias, if there were any, in the ligand set would be reduced [9]. An example of the importance of filtering the input data can be seen in the MTORC1 ligand set (Figure 2) included in our recently introduced LIT-PCBA data collection [1], comprising the molecules tested for an inhibitory activity towards the mTORC1 signaling pathway, targeting the human serine/threonine-protein kinase mTOR.
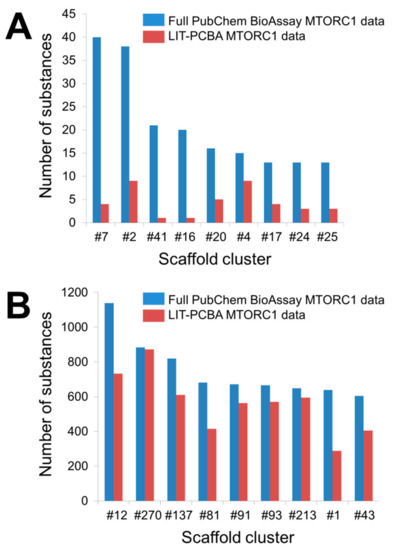
Figure 2. Number of substances falling into each scaffold cluster that includes more than 10 true active molecules (A) or 600 true inactive molecules (B). Bemis-Murcko frameworks derived from the input molecules were first created by trimming each active and each inactive separately with Pipeline Pilot 19.1.0.1964 [21][22]. A hierarchical scaffold tree consisting of canonical SMILES (simplified molecular-input line-entry system) strings that represent the rings, linkers, and double bonds in each molecule was next generated according to an iterative ring-trimming procedure described by Schuffenhauer et al. (2007) [23]. All ligands were then clustered based on the smallest scaffold at the root of the scaffold tree for each ligand. The number that follows each hash symbol indicated in this figure refers to the ordinal number of a scaffold cluster as issued by Pipeline Pilot. Details of all clusters can be found in the Supplementary Materials (Tables S3 and S4).
As to be expected, the full PubChem BioAssay data feature a larger number of scaffold clusters, with 59 clusters for the active set and 1151 clusters for the inactive set (against 41 and 1106 clusters in the LIT-PCBA active and inactive ligand sets, respectively). However, only 18 (out of 342; 5.26%) true actives possess unique scaffolds, meaning nearly 95% of all active substances in the full PubChem ligand set share chemical similarities with at least another active. Notably, nine clusters are reported to have more than 10 representatives (Figure 2A and Table S3). The pruned LIT-PCBA active ligand set, on the other hand, includes no cluster with over 10 members and 21 clusters (51.22%) with only one substance for each. This means nearly a quarter of the LIT-PCBA active molecules (over four times the value observed in the full PubChem set) possess unique scaffolds. Moreover, the number of ligands falling into each cluster in the filtered LIT-PCBA active set is greatly reduced in comparison to that of the unfiltered data (Figure 2A and Table S3). On the other hand, around 25% of PubChem molecules were deemed to have extreme physicochemical properties and were therefore discarded as the MTORC1 ligand set was constructed [1]. These observations suggest that (i) there is indeed significant chemical bias in the full PubChem active ligand composition, and (ii) the filtering steps that were applied to build the LIT-PCBA data collection helped reduce this bias by lowering the number of active substances sharing the same chemical features (thus avoiding the presence of too many molecules issued from the same chemotype) and by ruling out compounds that were too different from others (hence, preventing artificial enrichment). A similar conclusion can be drawn from the full PubChem inactive ligand set and the corresponding LIT-PCBA data (Figure 2B and Table S4). The benefit of filtering the PubChem ligands in reducing the chemical bias is again highlighted as the data sets undergo a subsequent unbiasing procedure using the previously described asymmetric validation embedding (AVE) method [24], which measures pairwise distances in the chemical space between molecules belonging to four sets of compounds (training actives, training inactives, validation actives, and validation inactives; training-to-validation ratio = 3) based on the ECFP4 fingerprints. A nearly zero overall bias value (0.001) was obtained from the LIT-PCBA MTORC1 ligand set after only seven iteration steps of the AVE genetic algorithm (GA) [1], while 16 GA iterations were necessary to bring the overall bias of the full PubChem data set down to 0.006. This denotes that the pruned LIT-PCBA ligands are much less biased, in terms of chemical features, than the complete PubChem molecules and confirms the necessity of detecting chemical bias in the composition of data deposited on PubChem BioAssay and removing them, if there were any, so that the data set is better adapted for further use.
The impact of filtering the PubChem BioAssay molecules on the subsequent retrospective screening performances can also be observed with the use of two in silico methods: a 2D similarity search using extended-connectivity ECFP4 fingerprints with Pipeline Pilot [22][25] (ligand-based) and molecular docking with Surflex-Dock (structure-based) [26]. Both data sets (the full PubChem data and the pruned LIT-PCBA MTORC1 ligands) underwent the same screening protocols using the two aforementioned programs, as described in our previous paper [1]. The screening performance was evaluated according to the EF1% (enrichment in true actives at a constant 1% false positive rate over random picking) values obtained by the “max-pooling approach”, taking into account all available PDB templates of the protein target (n = 11), while generating only one hit list that facilitated the post-screening assessments [1]. It was observed that both methods performed better on the full PubChem data than on the filtered LIT-PCBA ligand set (Table 1). Interestingly, the true actives that were retrieved along with the top 1% false positives belonged to the same scaffold clusters or to clusters that were similar to each other. Such observations reconfirm that (i) ligand-based and structure-based screening approaches tend to recognize compounds that share chemical features, and (ii) the chemical bias present in the complete PubChem data indeed leads to overoptimistic screening performances. This, again, highlights the importance of filtering the ensemble of molecules deposited on PubChem BioAssay prior to evaluating the virtual screening procedures—first, to reduce chemical bias in the composition of the data and, then, to avoid overestimating the real discriminatory accuracy of in silico methods.
Table 1. Retrospective screening performance of a 2D ECFP4 fingerprint similarity search with Pipeline Pilot and molecular docking with Surflex-Dock on the full PubChem BioAssay data and the pruned LIT-PCBA MTORC1 ligand set, demonstrated by the enrichment in true actives at a constant 1% false positive rate over random picking (EF1%) values and the numbers of true actives retrieved along with the top 1% false positives by the “max-pooling” approach.
| Data Sets | 2D ECFP4 Fingerprint Similarity Search | Molecular Docking | ||
|---|---|---|---|---|
| EF1% | Number of Retrieved Actives | EF1% | Number of Retrieved Actives | |
| Full PubChem data | 0.6 | 2 | 3.2 | 11 |
| LIT-PCBA MTORC1 data | 0.0 | 0 | 1.0 | 1 |
5. Potency Bias in the Composition of Active Ligand Sets
As of 30 April 2020, there were 1,067,719 small-molecule assays deposited on the PubChem BioAssay database, but only 240,999 of them (22.6%) yielded active substances with confirmed potency values. These values are provided in different terms (EC50, IC50, Kd, and Ki), and the threshold to distinguish true actives from true inactives varies from assay to assay, depending on the researchers who conducted the experiments. Some assays accept active substances with potency values above 100 µM (e.g., AIDs 1030, 1490, and 504847), even at the millimolar level (e.g., AIDs 1045 and 1047), while, in some others, several substances with even submicromolar potency are not deemed actives (e.g., AIDs 1221, 1224, and 1345010). It is therefore comprehensible that the potency range of true actives, as well as its distribution, is quite diverse across all assays of PubChem. As active molecules with high potency towards a biological target are easier to be picked by both ligand-based and structure-based virtual screening methods [1], ligand sets with too many actives whose potency values are in the submicromolar range are prone to overestimating the real accuracy of in silico screening. PubChem BioAssay data sets, especially those composed of highly potent true actives (potency below 1 µM), need to be filtered so that the so-called “potency bias” in the composition of their active ligand sets is reduced before further use.
An illustration of this point can be taken from the LIT-PCBA PPARG ligand set (27 true actives and 5211 true inactives) and the corresponding full PubChem BioAssay data (AID 743094; 78 true actives and 8532 true inactives) comprising small molecules that were tested for an agonistic activity on the peroxisome proliferator-activated receptor gamma (PPARg) signaling pathway [1]. The number of true actives with high potency (EC50 < 1 µM) in the complete PubChem data is 19, nearly three times higher than that of the pruned LIT-PCBA ligand set (n = 7). Upon carrying out a 2D similarity search with Pipeline Pilot using ECFP4 fingerprints and ten structurally diverse crystallographic PPARg agonists randomly chosen from 138 available structures on the Protein Data Bank as templates, it was observed that, as expected, the screening protocol managed to retrieve more highly potent true actives from the full data set than from the filtered ligand set in 70% of the cases (Figure 3). Moreover, the “max-pooling” approach, when applied to the complete PubChem data, selected seven highly potent actives among the top 1% ranked molecules, seven times higher than the amount obtained from LIT-PCBA. Among them, four even had potency values below 0.1 µM. The same screening method, on the other hand, failed to retrieve any true actives with EC50 < 0.1 µM from the pruned PPARG data. The screening performance observed on the full ligand set was, as a result, better than that obtained after ligand-filtering, as the EF1% value was nearly twice higher than that received with LIT-PCBA ligands. This reconfirms that in silico screening procedures tend to recognize molecules with high potency towards a protein target, and the presence of too many highly potent ligands in the data likely leads to a better screening performance. It is therefore recommended that one should filter the ensemble of PubChem BioAssay ligands to ensure that there are not too many true actives with high potency that remain, in order to avoid possible “potency bias” in the data set and the subsequent overestimation of in silico methods’ discriminatory power.
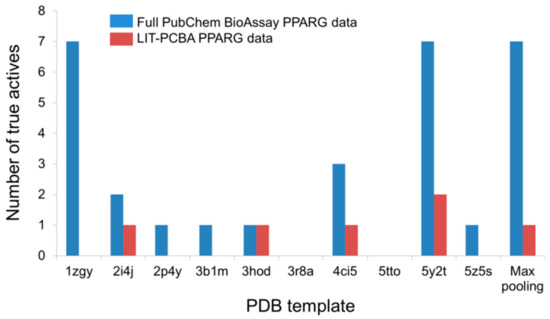
Figure 3. The number of highly potent true actives (EC50 < 1 µM) retrieved among the top 1% ranked molecules by a 2D ECFP4 fingerprint similarity search from the full PubChem BioAssay data and the corresponding LIT-PCBA PPARG ligand set after ligand-filtering. Ten known crystallographic PPARg agonists were randomly chosen as templates from 138 available structures on the Protein Data Bank.
6. Processing Input Structures Prior to Virtual Screening
PubChem BioAssay ligands, as deposited on the database, can be downloaded either as SMILES strings [27] or in 2D SDF (spatial data file) format [28] and are therefore, in general, not yet ready to be directly employed as the input for most in silico screening protocols (except for 1D or 2D ligand-based approaches). A rigorous ligand-processing procedure is thus necessary to afford ready-to-use structures for virtual screening. This process concerns a wide range of aspects inherent in the three-dimensional structural formula of a molecule, including atomic coordinates in the 3D space, a formal charge assigned on each atom, the presence of different protonation states and tautomeric shifts that slightly alter the structure, and the representation of undefined stereocenters or flexible rings, as well as the existence of multiple conformations and/or configurations [29]. Various studies have concluded that database-processing has indeed an impact on the screening performance; some processing stages are even indispensable to certain programs [29][30][31][32]. Kellenberger et al. (2004) [31], Perola and Charifson (2004) [32], and Cummings et al. (2007) [29] pointed out that the initial conformation and orientation in the 3D space of a molecule, which are determined based on details featured in the original SMILES string, may significantly affect the final enrichment output by a docking program. The performances of structure-based screening methods whose scoring functions rely on ligand-receptor interactions [33][34] may be sensitive to a change in the explicit hydrogen assignment or protonation states, as the positions of hydrogen-bonding groups and proton-carrying atoms are crucial to properly detecting intermolecular hydrogen bonds and ionic interactions, respectively [29][35]. While a generation of correct multiple conformers for a molecule is not imperative when it comes to carrying out docking with GOLD [36] or Surflex-Dock [26], this step has, in fact, a pivotal role in the 3D shape similarity search using ROCS (OpenEye) [37]. The examples mentioned above denote that good in silico screening outcomes do require the careful treatment of input ligand sets, and a thorough investigation of different data-processing procedures with commonly used programs (e.g., Protoss [38], Corina [39], MOE [40], Sybyl [41], and Daylight [42]) is thus recommended. If it is possible (if the data size is not too large), one should check each output structure by hand to ensure that the assigned atom types, bond types, stereochemical properties, and protonation states are correct before further use. This also applies to the protein structure preparation prior to screening, as structural features of the protein target, especially those of the binding site, are of indisputable importance to the structure-based virtual screening performance.
7. Conclusions
Retrieving experimental PubChem BioAssay data to construct novel data sets for virtual screening evaluations helps avoid assuming false negatives among inactive ligands, which is a problem inherent in artificially developed data collections. However, there remain several issues regarding assay selection, false active molecules, chemical bias, and potency bias, as well as data curation, which are worth noticing prior to employing PubChem input for database-designing purposes. To the best of our knowledge, there have been several publicly available data sets that were constructed from the data deposited on this repository, but the quantity is not yet considerable, and there still exist some limitations in the design of these data collections. More efforts in this regard are recommended, with the points raised in this manuscript taken into account, in order to offer more realistic data sets suitable for validating both ligand-based and structure-based in silico screening procedures in the future. Of course, the herein proposed good practices should also be applied to proprietary bioactivity data.
References
- Tran-Nguyen, V.K.; Jacquemard, C.; Rognan, D. LIT-PCBA: An unbiased data set for machine learning and virtual screening. J. Chem. Inf. Model. 2020.
- Rohrer, S.G.; Baumann, K. Maximum unbiased validation (MUV) data sets for virtual screening based on PubChem bioactivity data. J. Chem. Inf. Model. 2009, 49, 169–184.
- Lindh, M.; Svensson, F.; Schaal, W.; Zhang, J.; Sköld, C.; Brandt, P.; Karlen, A. Toward a benchmarking data set able to evaluate ligand- and structure-based virtual screening using public HTS data. J. Chem. Inf. Model. 2015, 55, 343–353.
- Huang, N.; Shoichet, B.K.; Irwin, J.J. Benchmarking sets for molecular docking. J. Med. Chem. 2006, 49, 6789–6801.
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594.
- Bauer, M.R.; Ibrahim, T.M.; Vogel, S.M.; Boeckler, F.M. Evaluation and optimization of virtual screening workflows with DEKOIS 2.0—A public library of challenging docking benchmark sets. J. Chem. Inf. Model. 2013, 53, 1447–1462.
- Schierz, A.C. Virtual screening of bioassay data. J. Cheminformatics 2009, 1, 21.
- Butkiewicz, M.; Lowe, E.W.; Mueller, R.; Mendenhall, J.L.; Teixeira, P.L.; Weaver, C.D.; Meiler, J. Benchmarking ligand-based virtual high-throughput screening with the PubChem database. Molecules 2013, 18, 735–756.
- Lagarde, N.; Zagury, J.F.; Montes, M. Benchmarking data sets for the evaluation of virtual ligand screening methods: Review and perspectives. J. Chem. Inf. Model. 2015, 55, 1297–1307.
- Nim, S.; Lobato, L.G.; Moreno, A.; Chaptal, V.; Rawal, M.K.; Falson, P.; Prasad, R. Atomic modelling and systematic mutagenesis identify residues in multiple drug binding sites that are essential for drug resistance in the major candida transporter Cdr1. Biochim. Biophys. Acta 2016, 1858, 2858–2870.
- Wang, Y.; Bolton, E.; Dracheva, S.; Karapetyan, K.; Shoemaker, B.A.; Suzek, T.O.; Wang, J.; Xiao, J.; Zhang, J.; Bryant, S.H. An overview of the PubChem BioAssay resource. Nucleic Acids Res. 2010, 38, D255–D266.
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249.
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740.
- Gilberg, E.; Jasial, S.; Stumpfe, D.; Dimova, D.; Bajorath, J. Highly promiscuous small molecules from biological screening assays include many pan-assay interference compounds but also candidates for polypharmacology. J. Med. Chem. 2016, 59, 10285–10290.
- Baell, J.B. Feeling nature’s PAINS: Natural products, natural product drugs, and pan assay interference compounds (PAINS). J. Nat. Prod. 2016, 79, 616–628.
- Capuzzi, S.J.; Muratov, E.N.; Tropsha, A. Phantom PAINS: Problems with the utility of alerts for pan-assay INterference CompoundS. J. Chem. Inf. Model. 2017, 57, 417–427.
- Kenny, P.W. Comment on the ecstasy and agony of assay interference compounds. J. Chem. Inf. Model. 2017, 57, 2640–2645.
- Baell, J.B.; Nissink, J.W. Seven year itch: Pan-assay interference compounds (PAINS) in 2017—Utility and limitations. ACS Chem. Biol. 2018, 13, 36–44.
- Hsieh, J.H. Accounting artifacts in high-throughput toxicity assays. Methods Mol. Biol. 2016, 1473, 143–152.
- Good, A.C.; Oprea, T.I. Optimization of CAMD techniques 3. Virtual screening enrichment studies: A help or hindrance in tool selection? J. Comput. Aided Mol. Des. 2008, 22, 169–178.
- Bemis, G.W.; Murcko, M.A. The properties of known drugs. 1. Molecular frameworks. J. Med. Chem. 1996, 39, 2887–2893.
- Dassault Systèmes, Biovia Corp. Available online: https://www.3dsbiovia.com/ (accessed on 1 April 2020).
- Schuffenhauer, A.; Ertl, P.; Roggo, S.; Wetzel, S.; Koch, M.A.; Waldmann, H. The scaffold tree, visualization of the scaffold universe by hierarchical scaffold classification. J. Chem. Inf. Model. 2007, 47, 47–58.
- Wallach, I.; Heifets, A. most ligand-based classification benchmarks reward memorization rather than generalization. J. Chem. Inf. Model. 2018, 58, 916–932.
- Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754.
- Jain, A.N. Surflex-dock 2.1: Robust performance from ligand energetic modeling, ring flexibility, and knowledge-based search. J. Comput. Aided Mol. Des. 2007, 21, 281–306.
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36.
- Dalby, A.; Nourse, J.G.; Hounshell, W.D.; Gushurst, A.K.; Grier, D.L.; Leland, B.A.; Laufer, J. Description of several chemical structure file formats used by computer programs developed at molecular design limited. J. Chem. Inf. Comput. Sci. 1992, 32, 244–255.
- Cummings, M.D.; Gibbs, A.C.; DesJarlais, R.L. Processing of small molecule databases for automated docking. Med. Chem. 2007, 3, 107–113.
- Knox, A.J.; Meegan, M.J.; Carta, G.; Lloyd, D.G. Considerations in compound database preparatio—“hidden” impact on virtual screening results. J. Chem. Inf. Model. 2005, 45(6), 1908–1919.
- Kellenberger, E.; Rodrigo, J.; Muller, P.; Rognan, D. Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins 2004, 57, 225–242.
- Perola, E.; Charifson, P.S. Conformational analysis of drug-like molecules bound to proteins: An extensive study of ligand reorganization upon binding. J. Med. Chem. 2004, 47, 2499–2510.
- Marcou, G.; Rognan, D. Optimizing fragment and scaffold docking by use of molecular interaction fingerprints. J. Chem. Inf. Model. 2007, 47, 195–207.
- Desaphy, J.; Raimbaud, E.; Ducrot, P.; Rognan, D. Encoding protein-ligand interaction patterns in fingerprints and graphs. J. Chem. Inf. Model. 2013, 53, 623–637.
- Polgar, T.; Keserue, G.M. Ensemble docking into flexible active sites. critical evaluation of FlexE against JNK-3 and β-secretase. J. Chem. Inf. Model. 2006, 46, 1795–1805.
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748.
- Hawkins, P.C.; Skillman, A.G.; Nicholls, A. Comparison of shape-matching and docking as virtual screening tools. J. Med. Chem. 2007, 50, 74–82.
- Bietz, S.; Urbaczek, S.; Schulz, B.; Rarey, M. Protoss: A holistic approach to predict tautomers and protonation states in proteinligand complexes. J. Cheminformatics 2014, 6, 12.
- Molecular Networks Gmbh. Available online: https://www.mn-am.com/ (accessed on 30 April 2020).
- Molecular Operating Environment. Available online: https://www.chemcomp.com/Products.htm (accessed on 1 May 2020).
- Sybyl-X Molecular Modeling Software Packages, Version 2.0; TRIPOS Associates, Inc.: St. Louis, MO, USA, 2012.
- Daylight Chemical Information Systems. Available online: https://www.daylight.com/ (accessed on 1 May 2020).

