Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marek Postula | + 2468 word(s) | 2468 | 2021-06-21 10:53:32 | | | |
| 2 | Lily Guo | Meta information modification | 2468 | 2021-06-30 03:05:09 | | | | |
| 3 | Lily Guo | Meta information modification | 2468 | 2021-06-30 03:05:45 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Postula, M. Atherosclerotic Cardiovascular Diseases. Encyclopedia. Available online: https://encyclopedia.pub/entry/11488 (accessed on 23 July 2026).
Postula M. Atherosclerotic Cardiovascular Diseases. Encyclopedia. Available at: https://encyclopedia.pub/entry/11488. Accessed July 23, 2026.
Postula, Marek. "Atherosclerotic Cardiovascular Diseases" Encyclopedia, https://encyclopedia.pub/entry/11488 (accessed July 23, 2026).
Postula, M. (2021, June 29). Atherosclerotic Cardiovascular Diseases. In Encyclopedia. https://encyclopedia.pub/entry/11488
Postula, Marek. "Atherosclerotic Cardiovascular Diseases." Encyclopedia. Web. 29 June, 2021.
Copy Citation
Atherosclerotic cardiovascular diseases (ASCVD) are the major cause of mortality worldwide. Despite the continuous progress in ASCVD therapy, the residual risk persists beyond the management of traditional risk factors. Several infections including Helicobacter pylori infection, periodontal disease, and viral infections are associated with the increased risk of ASCVD, both directly by damage to the heart muscle and vasculature, and indirectly by triggering a systemic proinflammatory state. Hence, beyond the optimal management of the traditional ASCVD risk factors, infections should be considered as an important non-classical risk factor to enable early diagnosis and appropriate treatment.
atherosclerosis
cardiovascular disease
infections
outcomes
risk factors
1. Introduction
Cardiovascular diseases (CVD) are the leading cause of mortality worldwide, accounting for an estimated 17.9 million cases of death per year [1]. Among them, the largest group is atherosclerotic CVD (ASCVD). Despite the continuous progress in ASCVD therapy, the residual risk persists beyond the management of traditional risk factors. Hence, there is still a need for a better comprehension of pathophysiological processes underlying ASCVD [2][3]. In addition to traditional risk factors such as smoking, visceral adiposity, diabetes, hypertension, and dyslipidemia, chronic inflammation is one of the well-established non-classical risk factors of atherosclerosis [4][5]. Prolonged exposure to oxidative stress leads to the activation of multiple cell types involved in inflammation, including leukocytes, platelets, endothelial cells, and smooth muscle cells. Cellular mediators released from these cells induce and sustain the chronic inflammation of the vessel wall, which is a crucial pathophysiological mechanism of atherosclerosis [6]. Vessel wall inflammation is also due to impaired production or activity of nitric oxide (NO), which leads to endothelial dysfunction at an early stage of atherosclerosis. Impaired NO release along with increased NO degradation leads to an imbalance between vasoconstriction and vasodilation and initiates numerous mechanisms that promote and exacerbate atherosclerosis [7].
Since infections are inevitably associated with activation of immune response and inflammatory processes, the relation between ASCVD and infections has been extensively studied. There is evidence that particular infections are a risk factor for the development and/or aggravation of ASCVD. In particular, the association of ASCVD with periodontal disease, Helicobacter pylori (H. pylori), Cytomegalovirus (CMV), pneumonia, Human immunodeficiency virus (HIV), Herpes simplex virus (HSV), and most recently severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) infection has been described [8][9][10][11][12][13][14]. However, the pathomechanism of these associations is not entirely clear and hitherto research has provided controversial results. Considering that ASCVD are the major cause of death in high-income countries, whereas infections (a potentially modifiable risk factor) are the major cause of death in middle- and low-income countries [15], it is crucial to determine both the pathophysiological associations and the possible advantages of infection treatment in the course of ASCVD. This may have a major effect not only on global human health but also on the healthcare costs related to infections and ASCVD. Here, we summarized the currently available evidence on the role of infections in ASCVD development, considering (i) the role of inflammation in ASCVD, (ii) the impact of particular infections and pathogens (Helicobacter pylori, periodontal disease, pneumonia, CMV, HCV, HIV, HSV, and most recently SARS-CoV-2) on the development and progression of ASCVD and (iii) possible therapeutic implications, focusing on the anti-inflammatory and anti-infective efficacy of statins, P2Y12 inhibitors, angiotensin-converting enzyme inhibitors (ACE-I) and angiotensin receptor blockers (ARBs), colchicine, anti-cytokine drugs, and methotrexate.
2. The Role of Inflammation in Cardiovascular Diseases
Atherosclerosis is an inflammatory disease that underlies ASCVD and related complications, including acute coronary syndrome, ischemic stroke, and peripheral artery disease [6]. Cells involved in the development of atherosclerosis include macrophages/foam cells, endothelial cells, smooth muscle cells (SMC), T-lymphocytes, and platelets. In addition, extracellular vesicles (EVs) and microRNAs (miRNAs) released from these cells induce and sustain atherosclerotic processes in the vessel wall. The pathophysiology of atherosclerotic plaque formation is shown in Figure 1.
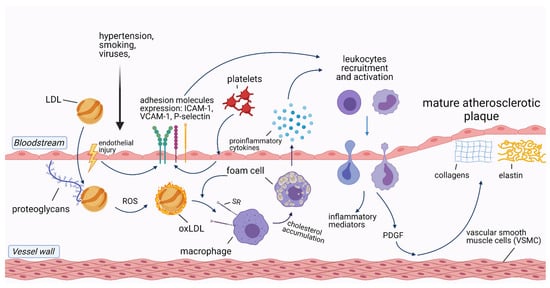
Figure 1. Pathophysiology of atherosclerotic plaque formation. Abbreviations: ICAM-1—intercellular adhesion molecule-1; LDL—low-density lipoprotein; oxLDL—oxidized LDL; PDGF—platelet-derived growth factor; ROS—reactive oxygen species; SR—scavenger receptor; VCAM-1—vascular cell adhesion protein 1. Created with Biorender.com, licensed version.
Accumulation of circulating low-density lipoproteins (LDL) in the subendothelial layer of the arteries is the initial stage in the development of atherosclerosis. Following endothelial injury, LDL adhere to extracellular matrix proteins exposed by the activated endothelial cells [16]. As LDL is trapped in the vascular wall, it undergoes oxidation by locally secreted reactive oxygen species (ROS), being transformed into oxidized LDL (ox-LDL) [17]. Ox-LDL, along with native LDL, are taken up by macrophages via scavenger receptors (SR), leading to macrophage transformation into foam cells [18]. Both macrophages and foam cells release a variety of proinflammatory cytokines such as soluble CD40 ligand, interleukin-1 (IL-1), IL-3, IL-6, IL-8, IL-18, and tumor necrosis factor-alpha (TNF-α) [19]. IL-1β, IL-6, and TNF-α stimulate the liver production of C-reactive protein (CRP), the most common inflammatory biomarker [20].
Besides macrophages/foam cells, endothelial cells are also involved in atherosclerotic plaque formation. After exposure to damaging factors, such as dyslipidemia, smoking, hypertension, or viruses, endothelial cells upregulate the transcription of nuclear factor-κB (NF-κB) and expose molecules that enhance leukocyte adhesion to the endothelium, such as P-selectin, E-selectin, vascular cell adhesion molecule-1 (VCAM-1), and intercellular adhesion molecule-1 (ICAM-1) [21]. Together with ox-LDL, activated leukocytes migrate through the injured endothelial layer to the intimal layer of the vessel wall, where they produce inflammatory mediators which sustain the inflammatory process [6].
Likewise, arterial SMC respond to chemoattractants produced by activated leukocytes, such as platelet-derived growth factor, which results in SMC migration from the tunica media of the artery wall to the intimal layer. There, SMC produce collagen, elastin, and glycosaminoglycans, which are the principal components of mature atherosclerotic plaques [19].
T-lymphocytes play a dual role in atherosclerosis. Regulatory T-lymphocytes (Tregs) which produce transforming growth factor β (TGF-β) and IL-10, as well as Th-17 lymphocytes that secrete IL-17, have a protective effect, promoting stabilization of atherosclerotic plaques due to the expansion of the fibrous cap [22]. In contrast, Th1 lymphocytes promote atherosclerosis by exuding proinflammatory components such as interferon-γ and TNF-α [23].
Platelets have an underappreciated role in atherogenesis. One of the mechanisms of their contribution is through their interactions with endothelial cells which result in the release of chemokines and expression of adhesion molecules (E-selectin, P-selectin, VCAM-1, ICAM-1). This is then followed by IL-1β secretion leading to the recruitment of immune cells [24]. Furthermore, platelets are able to participate in the oxidization of LDL and to promote LDL uptake by macrophages, thus contributing to the development of atherosclerosis [25].
The nucleotide-binding oligomerization domain-like receptors, pyrin domain-containing 3 (NLRP3) inflammasome, which is a component of the innate immune system, has been linked to several inflammatory disorders such as periodic cryopyrin syndromes, Alzheimer’s disease, diabetes, atherosclerosis, and more recently atrial fibrillation [26][27]. The NLRP3 inflammasome is responsible for caspase-1 activation and the secretion of proinflammatory cytokines IL-1β and IL-18 during microbial infection and cellular damage thus can contribute to ASCVD development [26].
Cells involved in the development of atherosclerosis secrete EVs and miRNAs that modulate inflammation of the blood vessel wall [18]. EVs are lipid membrane particles that are naturally released from cells into blood and other body fluids, which transport biomolecules between cells [28]. Multiple EVs subtypes are associated with pathophysiological processes underlying atherosclerosis and CVD, such as endothelial dysfunction, accumulation of lipids in the vessel wall, migration of immune cells, calcification of atherosclerotic plaques, and plaque rupture [29]. Moreover, EVs are involved in the adverse myocardial remodeling following acute myocardial infarction (AMI), making them not only biomarkers but also therapeutic targets in CVD [30][31]. The role of EVs in atherosclerotic development and progression has been summarized elsewhere [32].
MiRNAs are small, non-coding RNAs that regulate post-transcriptional gene expression [33]. For example, miR-181a-5p and its passenger strand miR-181a-3p act as negative post-transcriptional regulators of the NF-κB signaling pathway, thus contributing to inflammation. Restoring the expression of these miRNAs retarded not only the inflammation but also the progression of atherosclerosis in apoE−/− mice [34]. Additionally, miR-30c and miR-126p had a protective effect on ASCVD. MiR-30c was associated with hyperlipidemia and atherosclerosis reduction in the Western diet-fed mice, while miR-126p limited atherosclerosis by suppressing Notch1 inhibitor delta-like-1 homolog in Mir126−/− mice [35][36]. Conversely, miR-143 and miR-145 were characterized as vascular SMC physiology regulators, which are involved in SMC proliferation, migration, and apoptosis. miR-19b, miR-33, miR-92a, miR-144-3p, miR-155, and miR-342-5p were also shown to promote atherosclerosis [37][38][39][40][41][42]. Therefore, it is suggested that the dysregulation of miRNAs expression is associated with ASCVD development and progression [43]. Similar to EVs, miRNAs are also involved in post-infarction myocardial repair and could be used to design novel therapies to prevent and/ or reverse myocardial damage [44][45].
Discussing the role of inflammation in the development of atherosclerosis, good models of systemic inflammation are patients with Chronic Inflammatory Rheumatic diseases (CIRD) such as rheumatoid arthritis (RA) or systemic lupus erythematosus (SLE). Patients with CIRD are at increased risk of accelerated atherosclerosis compared with a healthy population [46]. Moreover, therapy with anti-inflammatory drugs such as anti-TNF or anti-IL-1 significantly reduces the risk, pointing to inflammation as the cause of the excessive risk of ASCVD [47].
Altogether, inflammatory processes involved in ASCVD are complex and require further investigation. Accumulating evidence shows that not only classical triggers of endothelial injury and inflammation but also infections may trigger and aggravate ASCVD. It remains unclear whether the mechanism by which infections affect atherosclerosis is similar to that in CIRD or whether additional factors play a role. There are also no direct comparisons between CIRD and infectious diseases in terms of ASCVD risk, thereby raising a need for additional research on this topic. However, the next paragraph focuses on infections as a risk for ASCVD.
3. Infections and Atherosclerotic Cardiovascular Diseases
The mechanisms underlying the increased risk of ASCVD in course of infections and discussed in this paragraph are summarized in Figure 2.
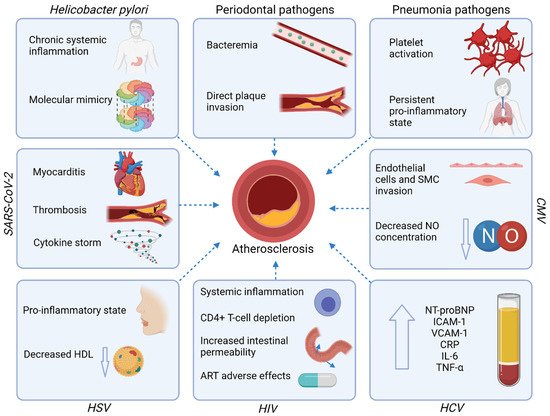
Figure 2. Mechanisms underlying the increased risk of ASCVD in course of infections. Abbreviations: ART—antiretroviral therapy; CMV—Cytomegalovirus; CRP—C-reactive protein; HCV—Hepatitis-C virus; HDL—high-density lipoprotein; HIV—Human immunodeficiency virus; HSV—Herpes simplex virus; ICAM-1—Intercellular adhesion molecule-1; IL—interleukin; NO—nitric oxide; NT-proBNP—N-terminal pro-B-type natriuretic peptide; SARS-CoV-2—Severe acute respiratory syndrome coronavirus-2; SMC—Smooth muscle cells; TNF-α—tumor necrosis factor-alpha; VCAM-1—Vascular cell adhesion protein 1. Created with Biorender.com, licensed version.
4. Therapeutic Implications
Regarding the excessive risk of ASCVD related to infections, it is crucial to establish the effective prevention and treatment of both entities. Here, we focus on the currently available strategies. However, since the residual risk related to chronic infections remains an important cardiovascular risk factor, there is an ongoing search for new therapeutics with the anti-inflammatory and/or anti-infective modes of action. This is particularly important given the strong literature reports of the ineffectiveness of anti-infective drugs, including antibiotics in the prevention of ASCVD [48]. The anti-inflammatory and/or anti-infective mechanisms of currently available drugs that may affect residual risk are shown in Figure 3. A summary of clinical studies evaluating the cardiovascular outcomes in patients receiving anti-inflammatory drugs is shown in Table 1.
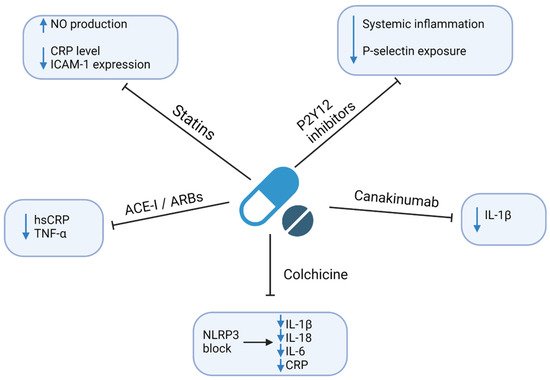
Figure 3. Anti-inflammatory and/or anti-infective mechanisms of currently available drugs. Abbreviations: ACEI—angiotensin-converting enzyme inhibitor; ARBs—angiotensin II receptor blocker; CRP—C-reactive protein; hsCRP—high-sensitivity CRP; ICAM-1—Intercellular Adhesion Molecule 1; IL—interleukin; NLRP3—nucleotide-binding oligomerization domain-like receptors, pyrin domain-containing 3; NO—nitric oxide; TNF-α—tumor necrosis factor-alpha. Created with Biorender.com, licensed version.
Table 1. Summary of clinical studies evaluating the cardiovascular outcomes in patients receiving anti-inflammatory drugs.
| Authors | Therapy | Mechanism of Action | Information about Study | Outcomes | Effect |
|---|---|---|---|---|---|
| Ridker et al., 2009 [49] | Rosuvastatin vs. placebo | HMG-CoA inhibitor, pleiotropic effects | A randomized, double-blind, placebo-controlled trial including 15,548 initially healthy men and women | Cardiovascular death, non-fatal stroke, non-fatal AMI, hospitalization due to unstable angina, revascularization | ↓ risk of adverse outcomes (HR = 0.35; 95% CI: 0.23–0.54; p < 0.0001) |
| Thomas et al., 2015 [50] | Ticagrelor vs. clopidogrel vs. placebo | Inhibition of P2Y12 receptor | Randomized injection of E. coli endotoxins to 30 healthy volunteers (10-ticagrelor, 10-clopidogrel, 10-placeboes) | Concentrations of inflammatory biomarkers | Ticagrelor and clopidogrel: ↓ IL6, TNF-α, CCL2 Only ticagrelor: ↓ G-CSF, IL-8; ↑ IL-10; ↔ hsCRP |
| McMurray et al., 2006 [51] | Valsartan vs. captopril | ARB or ACE inhibition | Randomized 14,703 high-risk patients with acute MI to receive captopril or valsartan or the combination of the two | All-cause mortality, cardiovascular mortality, non-fatal cardiovascular events | ↓ risk of adverse outcomes; similar effect of ARBs and ACE-I (HR = 0.97; 95% CI:0.91–1.03; p = 0.286) |
| Tardif et al., 2020 [52] | Colchicine 0.5 mg daily vs. placebo | NLRP3 inflammasome inhibitor | A randomized, double-blind, placebo-controlled trial including 4745 patients with recent AMI (~2 weeks before) | Cardiovascular death, resuscitated cardiac arrest, AMI, stroke, coronary revascularization | ↓ risk of adverse outcomes (HR = 0.77; 95% CI: 0.61–0.96; p = 0.02) |
| Nidorf et al., 2019 [53] | Colchicine 0.5 mg daily vs. placebo | NLRP3 inflammasome inhibitor | A randomized, placebo-controlled, double-blind trial including 5522 patients with chronic coronary syndrome | Cardiovascular death, MI, ischemic stroke, coronary revascularization | ↓ risk of adverse outcomes (HR = 0.69; 95% CI: 0.57–0.83; p < 0.001) |
| Nidorf et al., 2013 [54] | Colchicine 0.5 mg daily vs. placebo | NLRP3 inflammasome inhibitor | A prospective, randomized, observer-blinded, placebo-controlled clinical trial including 532 patients with stable coronary disease | Acute coronary syndrome, out-of-hospital cardiac arrest, ischemic stroke | ↓ risk of adverse outcomes (HR = 0.33; 95% CI: 0.18–0.59; p < 0.001) |
| Ridker et al., 2017 [55] | Canakinumab 150 mg every 3 months vs. placebo | Monoclonal anti-IL-1β antibody | A randomized, double-blind, placebo-controlled trial including 10,061 patients with previous AMI and hsCRP ≥ 2 mg/L | Non-fatal myocardial infarction, nonfatal stroke, cardiovascular death | ↓ risk of adverse outcomes HR = 0.85 (95% CI: 0.74–0.98; p = 0.021) |
| Greenberg et al., 2010 [56] | TNF-α antagonists vs. DMARDs | TNF-α inhibition | A longitudinal cohort study of 10,156 rheumatoid arthritis patients enrolled in the US-based CORRONA database | Non-fatal MI, transient ischemic attack, stroke, cardiovascular death | ↓ risk of adverse outcomes by TNF-α (HR = 0.39; 95% CI 0.19–0.82) |
| Ridker et al., 2019 [57] | Methotrexate 15–20 mg/week vs. placebo | Antimetabolite, immune-system suppressant | A randomized, double-blind, placebo-controlled trial including 4786 patients with previous MI or multivessel coronary disease, additionally with type 2 diabetes or metabolic syndrome | Nonfatal MI, nonfatal stroke, cardiovascular death, unstable angina | ↔ adverse outcomes (HR = 0.96; 95% CI: 0.79–1.16; p = 0.67) ↔ hsCRP, IL-1β, IL-6 ↑ ALT, AST |
5. Conclusions
Several infections including H. pylori infection, periodontal disease, and viral infections are associated with the increased risk of ASCVD, both directly by damage to the heart muscle and vasculature, and indirectly by triggering a systemic proinflammatory state. Hence, beyond the optimal management of the traditional ASCVD risk factors, infections should be considered as an important non-classical cardiovascular risk factor to enable early diagnosis and appropriate treatment. Numerous drugs routinely administered in patients with ASCVD including statins, P2Y12 inhibitors, and ACEi/ARBs were shown to have anti-inflammatory and/or anti-infective properties. However, no randomized controlled trials have hitherto evaluated their efficacy in patients with infective diseases. Novel strategies aiming at residual risk reduction have been proposed, including colchicine, anti-cytokine drugs, and methotrexate. However, more evidence regarding the risk-to-benefit ratio and cost-efficiency of these drugs is required before their clinical routine implementation in patients with infection-related high residual risk of CVD. Additional research is also needed on new therapeutic strategies, such as, for example, anti-atherosclerotic vaccines that already exist in the literature and where the biggest challenge is finding the appropriate antigen to immunize [48]. Nevertheless, further progress in understanding and therapeutic application of the relationship between infections/chronic inflammation and ASCVD requires well-designed, large studies with long-term follow-up.
References
- WHO Data. Available online: (accessed on 20 March 2021).
- Mihaylova, B.; Emberson, J.; Blackwell, L.; Keech, A.; Simes, J.; Barnes, E.H.; Voysey, M.; Gray, A.; Collins, R.; Baigent, C.; et al. The Effects of Lowering LDL Cholesterol with Statin Therapy in People at Low Risk of Vascular Disease: Meta-Analysis of Individual Data from 27 Randomised Trials. Lancet 2012, 380, 581–590.
- Kaasenbrood, L.; Boekholdt, S.M.; Van Der Graaf, Y.; Ray, K.K.; Peters, R.J.G.; Kastelein, J.J.P.; Amarenco, P.; Larosa, J.C.; Cramer, M.J.M.; Westerink, J.; et al. Distribution of Estimated 10-Year Risk of Recurrent Vascular Events and Residual Risk in a Secondary Prevention Population. Circulation 2016, 134, 1419–1429.
- Lechner, K.; von Schacky, C.; McKenzie, A.L.; Worm, N.; Nixdorff, U.; Lechner, B.; Kränkel, N.; Halle, M.; Krauss, R.M.; Scherr, J. Lifestyle Factors and High-Risk Atherosclerosis: Pathways and Mechanisms beyond Traditional Risk Factors. Eur. J. Prev. Cardiol. 2020, 27, 394–406.
- Corrado, E.; Novo, S. Role of Inflammation and Infection in Vascular Disease. Acta Chir. Belg. 2005, 105, 567–579.
- Alfarisi, H.A.H.; Mohamed, Z.B.H.; Ibrahim, M. Bin. Basic Pathogenic Mechanisms of Atherosclerosis. Egypt. J. Basic Appl. Sci. 2020, 7, 116–125.
- Davignon, J.; Ganz, P. Role of Endothelial Dysfunction in Atherosclerosis. Circulation 2004, 109 (Suppl. 23).
- Leishman, S.J.; Do, H.L.; Ford, P.J. Cardiovascular Disease and the Role of Oral Bacteria. J. Oral Microbiol. 2010, 2, 5781.
- Xu, Z.; Li, J.; Wang, H.; Xu, G. Helicobacter Pylori Infection and Atherosclerosis: Is There a Causal Relationship? Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 2293–2301.
- Lebedeva, A.M.; Shpektor, A.V.; Vasilieva, E.Y.; Margolis, L.B. Cytomegalovirus Infection in Cardiovascular Diseases. Biochemistry 2018, 83, 1437–1447.
- Restrepo, M.I.; Reyes, L.F. Pneumonia as a Cardiovascular Disease. Respirology 2018, 23, 250–259.
- Berquist, V.; Hoy, J.F.; Trevillyan, J.M. Contribution of Common Infections to Cardiovascular Risk in HIV-Positive Individuals. AIDS Rev. 2017, 19, 72–80.
- Wu, Y.P.; Sun, D.D.; Wang, Y.; Liu, W.; Yang, J. Herpes Simplex Virus Type 1 and Type 2 Infection Increases Atherosclerosis Risk: Evidence Based on a Meta-Analysis. BioMed Res. Int. 2016, 2016, 2630865.
- Dhakal, B.P.; Sweitzer, N.K.; Indik, J.H.; Acharya, D.; William, P. SARS-CoV-2 Infection and Cardiovascular Disease: COVID-19 Heart. Heart Lung Circ. 2020, 29, 973–987.
- WHO Data. Available online: (accessed on 2 May 2021).
- Williams, K.J.; Tabas, I. The Response-to-Retention Hypothesis of Early Atherogenesis. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 551–562.
- Hansson, G.K.; Hermansson, A. The Immune System in Atherosclerosis. Nat. Immunol. 2011, 12, 204–212.
- Golia, E.; Limongelli, G.; Natale, F.; Fimiani, F.; Maddaloni, V.; Pariggiano, I.; Bianchi, R.; Crisci, M.; D’Acierno, L.; Giordano, R.; et al. Inflammation and Cardiovascular Disease: From Pathogenesis to Therapeutic Target. Curr. Atheroscler. Rep. 2014, 16, 435.
- Libby, P.; Ridker, P.M. Inflammation and Atherothrombosis. From Population Biology and Bench Research to Clinical Practice. J. Am. Coll. Cardiol. 2006, 48, 33–46.
- Lorenzatti, A.J.; Servato, M.L. New Evidence on the Role of Inflammation in CVD Risk. Curr. Opin. Cardiol. 2019, 34, 418–423.
- Raggi, P.; Genest, J.; Giles, J.T.; Rayner, K.J.; Dwivedi, G.; Beanlands, R.S.; Gupta, M. Role of Inflammation in the Pathogenesis of Atherosclerosis and Therapeutic Interventions. Atherosclerosis 2018, 276, 98–108.
- Gisterå, A.; Robertson, A.K.L.; Andersson, J.; Ketelhuth, D.F.J.; Ovchinnikova, O.; Nilsson, S.K.; Lundberg, A.M.; Li, M.O.; Flavell, R.A.; Hansson, G.K. Transforming Growth Factor-β Signaling in T Cells Promotes Stabilization of Atherosclerotic Plaques through an Interleukin-17-Dependent Pathway. Sci. Transl. Med. 2013, 5, 18–23.
- Libby, P.; Hansson, G.K. Taming Immune and Inflammatory Responses to Treat Atherosclerosis. J. Am. Coll. Cardiol. 2018, 71, 173–176.
- Lordan, R.; Tsoupras, A.; Zabetakis, I. Platelet Activation and Prothrombotic Mediators at the Nexus of Inflammation and Atherosclerosis: Potential Role of Antiplatelet Agents. Blood Rev. 2021, 45, 100694.
- Gąsecka, A.; Rogula, S.; Szarpak, Ł.; Filipiak, K.J. LDL-Cholesterol and Platelets: Insights into Their Interactions in Atherosclerosis. Life 2021, 11, 39.
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328.
- Heijman, J.; Muna, A.P.; Veleva, T.; Molina, C.E.; Sutanto, H.; Tekook, M.; Wang, Q.; Abu-Taha, I.H.; Gorka, M.; Künzel, S.; et al. Atrial Myocyte NLRP3/CaMKII Nexus Forms a Substrate for Postoperative Atrial Fibrillation. Circ. Res. 2020, 127, 1036–1055.
- Van der Pol, E.; Böing, A.N.; Harrison, P.; Sturk, A.; Nieuwland, R. Classification, Functions, and Clinical Relevance of Extracellular Vesicles. Pharmacol. Rev. 2012, 64, 676–705.
- Hafiane, A.; Daskalopoulou, S.S. Extracellular Vesicles Characteristics and Emerging Roles in Atherosclerotic Cardiovascular Disease. Metabolism 2018, 85, 213–222.
- Gąsecka, A.; Pluta, K.; Solarska, K.; Rydz, B.; Eyileten, C.; Postula, M.; Van Der Pol, E.; Nieuwland, R.; Budnik, M.; Kochanowski, J.; et al. Plasma Concentrations of Extracellular Vesicles Are Decreased in Patients with Post-Infarct Cardiac Remodelling. Biology 2021, 10, 97.
- Gąsecka, A.; Van Der Pol, E.; Nieuwland, R.; Stępień, E. Extracellular Vesicles in Post-Infarct Ventricular Remodelling. Kardiol. Pol. 2018, 76, 69–76.
- Gasecka, A.; Nieuwland, R.; Siljander, P.R.M. Platelet-Derived Extracellular Vesicles. In Platelets, 4th ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 401–416.
- Wojciechowska, A.; Braniewska, A.; Kozar-Kamińska, K. MicroRNA in Cardiovascular Biology and Disease. Adv. Clin. Exp. Med. 2017, 26, 865–874.
- Su, Y.; Yuan, J.; Zhang, F.; Lei, Q.; Zhang, T.; Li, K.; Guo, J.; Hong, Y.; Bu, G.; Lv, X.; et al. MicroRNA-181a-5p and MicroRNA-181a-3p Cooperatively Restrict Vascular Inflammation and Atherosclerosis. Cell Death Dis. 2019, 10, 1–5.
- Soh, J. MicroRNA-30c Reduces Hyperlipidemia and Atherosclerosis by Decreasing Lipid Synthesis and Lipoprotein Secretion. Physiol. Behav. 2016, 176, 100–106.
- Schober, A.; Nazari-Jahantigh, M.; Wei, Y.; Bidzhekov, K.; Gremse, F.; Grommes, J.; Megens, R.T.A.; Heyll, K.; Noels, H.; Hristov, M.; et al. MicroRNA-126-5p Promotes Endothelial Proliferation and Limits Atherosclerosis by Suppressing Dlk1. Nat. Med. 2014, 20, 368–376.
- Lv, Y.C.; Tang, Y.Y.; Peng, J.; Zhao, G.J.; Yang, J.; Yao, F.; Ouyang, X.P.; He, P.P.; Xie, W.; Tan, Y.L.; et al. MicroRNA-19b Promotes Macrophage Cholesterol Accumulation and Aortic Atherosclerosis by Targeting ATP-Binding Cassette Transporter A1. Atherosclerosis 2014, 236, 215–226.
- Ouimet, M.; Ediriweera, H.; Afonso, M.S.; Ramkhelawon, B.; Singaravelu, R.; Liao, X.; Bandler, R.C.; Rahman, K.; Fisher, E.A.; Rayner, K.J.; et al. Microrna-33 regulates macrophage autophagy in atherosclerosis. Arterioscler. Thrombo. Vasc. Biol. 2017, 37, 1058–1067.
- Loyer, X.; Potteaux, S.; Vion, A.C.; Guérin, C.L.; Boulkroun, S.; Rautou, P.E.; Ramkhelawon, B.; Esposito, B.; Dalloz, M.; Paul, J.L.; et al. Inhibition of MicroRNA-92a Prevents Endothelial Dysfunction and Atherosclerosis in Mice. Circ. Res. 2014, 114, 434–443.
- Hu, Y.W.; Hu, Y.R.; Zhao, J.Y.; Li, S.F.; Ma, X.; Wu, S.G.; Lu, J.B.; Qiu, Y.R.; Sha, Y.H.; Wang, Y.C.; et al. An Agomir of MiR-144-3p Accelerates Plaque Formation through Impairing Reverse Cholesterol Transport and Promoting pro-Inflammatory Cytokine Production. PLoS ONE 2014, 9, e94997.
- Nazari-Jahantigh, M.; Wei, Y.; Noels, H.; Akhtar, S.; Zhou, Z.; Koenen, R.R.; Heyll, K.; Gremse, F.; Kiessling, F.; Grommes, J.; et al. MicroRNA-155 Promotes Atherosclerosis by Repressing Bcl6 in Macrophages. J. Clin. Investig. 2012, 122, 4190–4202.
- Wei, Y.; Nazari-Jahantigh, M.; Chan, L.; Zhu, M.; Heyll, K.; Corbalán-Campos, J.; Hartmann, P.; Thiemann, A.; Weber, C.; Schober, A. The MicroRNA-342-5p Fosters Inflammatory Macrophage Activation through an Akt1- and MicroRNA-155-Dependent Pathway during Atherosclerosis. Circulation 2013, 127, 1609–1619.
- Vacante, F.; Denby, L.; Sluimer, J.C.; Baker, A.H. The Function of MiR-143, MiR-145 and the MiR-143 Host Gene in Cardiovascular Development and Disease. Vascul. Pharmacol. 2019, 112, 24–30.
- Gozdowska, R.; Makowska, A.; Gąsecka, A.; Chabior, A.; Marchel, M. Circulating MicroRNA in Heart Failure—Practical Guidebook to Clinical Application. Cardiol Rev. 2020.
- Tomaniak, M.; Sygitowicz, G.; Filipiak, K.J.; Błaszczyk, O.; Kołtowski, L.; Gasecka, A.; Kochanowski, J.; Sitkiewicz, D. Dysregulations of MiRNAs and Galectin-3 May Underlie Left Ventricular Dilatation in Patients with Systolic Heart Failure. Kardiol. Pol. 2018, 76, 1012–1014.
- Arida, A.; Protogerou, A.D.; Kitas, G.D.; Sfikakis, P.P. Systemic Inflammatory Response and Atherosclerosis: The Paradigm of Chronic Inflammatory Rheumatic Diseases. Int. J. Mol. Sci. 2018, 19, 1890.
- Adawi, M.; Firas, S.; Blum, A. Rheumatoid Arthritis and Atherosclerosis. Isr. Med. Assoc. J. 2019, 21, 460–463.
- Pothineni, N.V.K.; Subramany, S.; Kuriakose, K.; Shirazi, L.F.; Romeo, F.; Shah, P.K.; Mehta, J.L. Infections, Atherosclerosis, and Coronary Heart Disease. Eur. Heart J. 2017, 38, 3195–3201.
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.; Genest, J.; Gotto, A.M.; Kastelein, J.J.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Reduction in C-Reactive Protein and LDL Cholesterol and Cardiovascular Event Rates after Initiation of Rosuvastatin: A Prospective Study of the JUPITER Trial. Lancet 2009, 373, 1175–1182.
- Thomas, M.R.; Outteridge, S.N.; Ajjan, R.A.; Phoenix, F.; Sangha, G.K.; Faulkner, R.E.; Ecob, R.; Judge, H.M.; Khan, H.; West, L.E.; et al. Platelet P2Y12 Inhibitors Reduce Systemic Inflammation and Its Prothrombotic Effects in an Experimental Human Model. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2562–2570.
- McMurray, J.; Solomon, S.; Pieper, K.; Reed, S.; Rouleau, J.; Velazquez, E.; White, H.; Howlett, J.; Swedberg, K.; Maggioni, A.; et al. The Effect of Valsartan, Captopril, or Both on Atherosclerotic Events after Acute Myocardial Infarction: An Analysis of the Valsartan in Acute Myocardial Infarction Trial (VALIANT). J. Am. Coll. Cardiol. 2006, 47, 726–733.
- Tardif, J.-C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505.
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.-F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in Patients with Chronic Coronary Disease. N. Engl. J. Med. 2020, 383, 1838–1847.
- Nidorf, S.M.; Eikelboom, J.W.; Budgeon, C.A.; Thompson, P.L. Low-Dose Colchicine for Secondary Prevention of Cardiovascular Disease. J. Am. Coll. Cardiol. 2013, 61, 404–410.
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131.
- Greenberg, J.D.; Kremer, J.M.; Curtis, J.R.; Hochberg, M.C.; Reed, G.; Tsao, P.; Farkouh, M.E.; Nasir, A.; Setoguchi, S.; Solomon, D.H. Tumour Necrosis Factor Antagonist Use and Associated Risk Reduction of Cardiovascular Events among Patients with Rheumatoid Arthritis. Ann. Rheum. Dis. 2011, 70, 576–582.
- Ridker, P.M.; Everett, B.M.; Pradhan, A.; Macfadyen, J.G.; Solomon, D.H.; Zaharris, E.; Mam, V.; Hasan, A.; Heart, N.; Rosenberg, Y.; et al. Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. N. Engl. J. Med. 2019, 380, 752–762.
More
Information
Subjects:
Cardiac & Cardiovascular Systems
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.7K
Revisions:
3 times
(View History)
Update Date:
29 Jan 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No