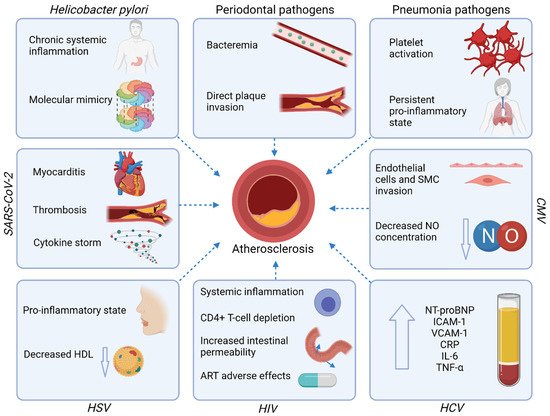
Atherosclerotic cardiovascular diseases (ASCVD) are the major cause of mortality worldwide. Despite the continuous progress in ASCVD therapy, the residual risk persists beyond the management of traditional risk factors. Several infections including Helicobacter pylori infection, periodontal disease, and viral infections are associated with the increased risk of ASCVD, both directly by damage to the heart muscle and vasculature, and indirectly by triggering a systemic proinflammatory state. Hence, beyond the optimal management of the traditional ASCVD risk factors, infections should be considered as an important non-classical risk factor to enable early diagnosis and appropriate treatment.
- atherosclerosis
- cardiovascular disease
- infections
- outcomes
- risk factors
1. Introduction
2. The Role of Inflammation in Cardiovascular Diseases
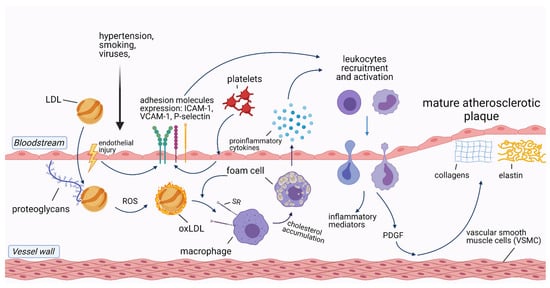
3. Infections and Atherosclerotic Cardiovascular Diseases
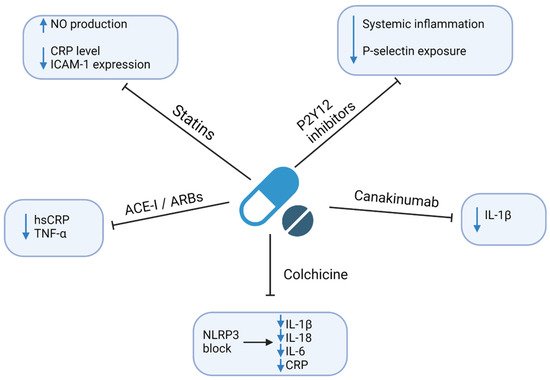
4. Therapeutic Implications
| Authors | Therapy | Mechanism of Action | Information about Study | Outcomes | Effect |
|---|---|---|---|---|---|
| Ridker et al., 2009 [111] | Rosuvastatin vs. placebo | HMG-CoA inhibitor, pleiotropic effects | A randomized, double-blind, placebo-controlled trial including 15,548 initially healthy men and women | Cardiovascular death, non-fatal stroke, non-fatal AMI, hospitalization due to unstable angina, revascularization | ↓ risk of adverse outcomes (HR = 0.35; 95% CI: 0.23–0.54; p < 0.0001) |
| Thomas et al., 2015 [112] | Ticagrelor vs. clopidogrel vs. placebo | Inhibition of P2Y12 receptor | Randomized injection of E. coli endotoxins to 30 healthy volunteers (10-ticagrelor, 10-clopidogrel, 10-placeboes) | Concentrations of inflammatory biomarkers | Ticagrelor and clopidogrel: ↓ IL6, TNF-α, CCL2 Only ticagrelor: ↓ G-CSF, IL-8; ↑ IL-10; ↔ hsCRP |
| McMurray et al., 2006 [113] | Valsartan vs. captopril | ARB or ACE inhibition | Randomized 14,703 high-risk patients with acute MI to receive captopril or valsartan or the combination of the two | All-cause mortality, cardiovascular mortality, non-fatal cardiovascular events | ↓ risk of adverse outcomes; similar effect of ARBs and ACE-I (HR = 0.97; 95% CI:0.91–1.03; p = 0.286) |
| Tardif et al., 2020 [114] | Colchicine 0.5 mg daily vs. placebo | NLRP3 inflammasome inhibitor | A randomized, double-blind, placebo-controlled trial including 4745 patients with recent AMI (~2 weeks before) | Cardiovascular death, resuscitated cardiac arrest, AMI, stroke, coronary revascularization | ↓ risk of adverse outcomes (HR = 0.77; 95% CI: 0.61–0.96; p = 0.02) |
| Nidorf et al., 2019 [115] | Colchicine 0.5 mg daily vs. placebo | NLRP3 inflammasome inhibitor | A randomized, placebo-controlled, double-blind trial including 5522 patients with chronic coronary syndrome | Cardiovascular death, MI, ischemic stroke, coronary revascularization | ↓ risk of adverse outcomes (HR = 0.69; 95% CI: 0.57–0.83; p < 0.001) |
| Nidorf et al., 2013 [116] | Colchicine 0.5 mg daily vs. placebo | NLRP3 inflammasome inhibitor | A prospective, randomized, observer-blinded, placebo-controlled clinical trial including 532 patients with stable coronary disease | Acute coronary syndrome, out-of-hospital cardiac arrest, ischemic stroke | ↓ risk of adverse outcomes (HR = 0.33; 95% CI: 0.18–0.59; p < 0.001) |
| Ridker et al., 2017 [117] | Canakinumab 150 mg every 3 months vs. placebo | Monoclonal anti-IL-1β antibody | A randomized, double-blind, placebo-controlled trial including 10,061 patients with previous AMI and hsCRP ≥ 2 mg/L | Non-fatal myocardial infarction, nonfatal stroke, cardiovascular death | ↓ risk of adverse outcomes HR = 0.85 (95% CI: 0.74–0.98; p = 0.021) |
| Greenberg et al., 2010 [118] | TNF-α antagonists vs. DMARDs | TNF-α inhibition | A longitudinal cohort study of 10,156 rheumatoid arthritis patients enrolled in the US-based CORRONA database | Non-fatal MI, transient ischemic attack, stroke, cardiovascular death | ↓ risk of adverse outcomes by TNF-α (HR = 0.39; 95% CI 0.19–0.82) |
| Ridker et al., 2019 [119] | Methotrexate 15–20 mg/week vs. placebo | Antimetabolite, immune-system suppressant | A randomized, double-blind, placebo-controlled trial including 4786 patients with previous MI or multivessel coronary disease, additionally with type 2 diabetes or metabolic syndrome | Nonfatal MI, nonfatal stroke, cardiovascular death, unstable angina | ↔ adverse outcomes (HR = 0.96; 95% CI: 0.79–1.16; p = 0.67) ↔ hsCRP, IL-1β, IL-6 ↑ ALT, AST |
5. Conclusions
This entry is adapted from the peer-reviewed paper 10.3390/jcm10122539