 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Giorgia Pallafacchina | + 4595 word(s) | 4595 | 2021-05-24 05:46:11 | | | |
| 2 | Bruce Ren | + 1744 word(s) | 6339 | 2021-06-28 03:04:57 | | |
Video Upload Options
The notion of the active role of mitochondria in the decoding and shaping of intracellular Ca2+ signals dates back at the end of the 19th century. However, the identity of the molecule(s) involved in Ca2+ ion transport into mitochondria remained elusive for decades. Only in the last ten years, the factors, and the relative coding genes, mediating Ca2+ entry in mitochondria started to be genetically and biochemically described. The gene for the pore-forming unit of the mitochondrial Ca2+ channel was discovered in 2011, and its product was named mitochondrial Ca2+ uniporter or MCU. The mitochondria Ca2+ uptake regulator 1 gene, MICU1, was cloned one year before, in 2010. The increasing interest of the scientific community towards mitochondrial Ca2+ signaling and metabolism in the subsequent years led to the identification of many other MCU components and to the description of their 3D structure and physiological role. Here, we will present a brief overview of the land marking discoveries in the history of mitochondrial Ca2+ studies.
1. Introduction
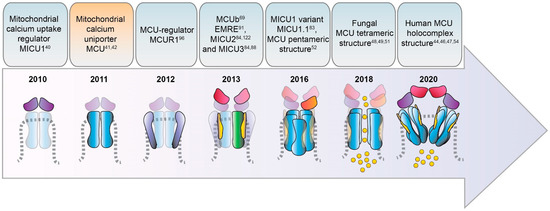
2. Timeline of MCU Identification
3. Discovery and Characterization of the MCU Complex Components
3.1. MCU
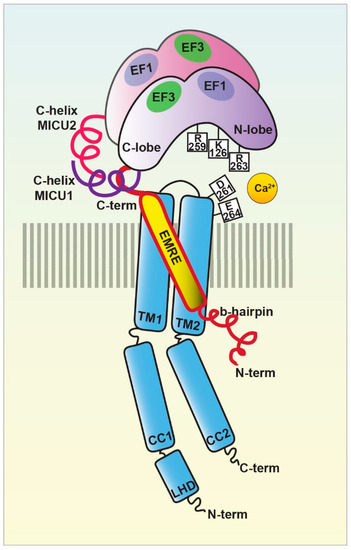
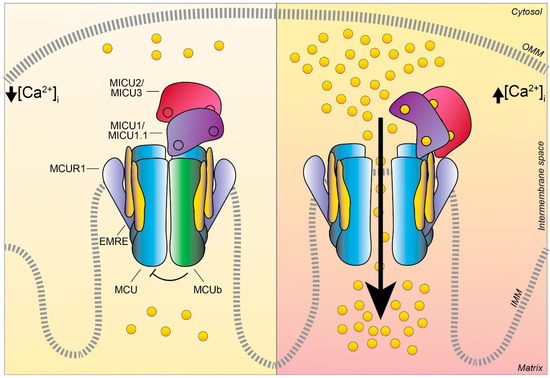
3.2. MCUb
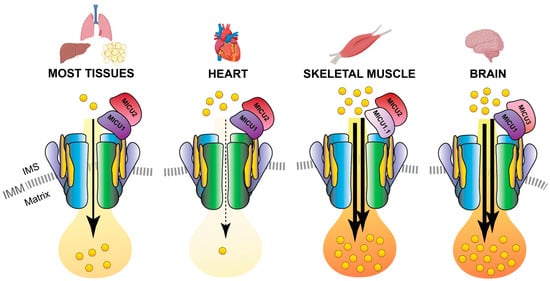
3.3. MICU1
3.4. MICU1.1
3.5. MICU2
3.6. MICU3
3.7. EMRE
References
- Raffaello, A.; Mammucari, C.; Gherardi, G.; Rizzuto, R. Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem. Sci. 2016, 41, 1035–1049.
- Clapham, D.E. Calcium Signaling. Cell 2007, 131, 1047–1058.
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21.
- Ringer, S. A third contribution regarding the Influence of the Inorganic Constituents of the Blood on the Ventricular Contraction. J. Physiol. 1883, 4, 222–225.
- Rizzuto, R.; Pozzan, T. Microdomains of Intracellular Ca2+: Molecular Determinants and Functional Consequences. Physiol. Rev. 2006, 86, 369–408.
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578.
- Denton, R.M.; Randle, P.J.; Martin, B.R. Stimulation by calcium ions of pyruvate dehydrogenase phosphate phosphatase. Biochem. J. 1972, 128, 161–163.
- Denton, R.M.; A Richards, D.; Chin, J.G. Calcium ions and the regulation of NAD+-linked isocitrate dehydrogenase from the mitochondria of rat heart and other tissues. Biochem. J. 1978, 176, 899–906.
- McCormack, J.G.; Denton, R.M. The effects of calcium ions and adenine nucleotides on the activity of pig heart 2-oxoglutarate dehydrogenase complex. Biochem. J. 1979, 180, 533–544.
- Territo, P.R.; Mootha, V.K.; French, S.A.; Balaban, R.S. Ca2+ activation of heart mitochondrial oxidative phosphorylation: role of the F0/F1-ATPase. Am. J. Physiol. Physiol. 2000, 278, C423–C435.
- Glancy, B.; Willis, W.T.; Chess, D.J.; Balaban, R.S. Effect of Calcium on the Oxidative Phosphorylation Cascade in Skeletal Muscle Mitochondria. Biochem. 2013, 52, 2793–2809.
- Quintana, A.; Pasche, M.; Junker, C.; Alansary, D.; Rieger, H.; Kummerow, C.; Nuñez, L.; Villalobos, C.; Meraner, P.; Becherer, U.; et al. Calcium microdomains at the immunological synapse: how ORAI channels, mitochondria and calcium pumps generate local calcium signals for efficient T-cell activation. EMBO J. 2011, 30, 3895–3912.
- Rasola, A.; Bernardi, P. The mitochondrial permeability transition pore and its involvement in cell death and in disease pathogenesis. Apoptosis 2007, 12, 815–833.
- Penna, E.; Espino, J.; De Stefani, D.; Rizzuto, R. The MCU complex in cell death. Cell Calcium 2018, 69, 73–80.
- Pinton, P.; Romagnoli, A.; Rizzuto, R.; Giorgi, C. Ca2+ signaling, mitochondria and cell death. Curr. Mol. Med. 2008, 8, 119–130.
- Heilbrunn, L.V.; Wiercinski, F.J. The action of various cations on muscle protoplasm. J. Cell. Comp. Physiol. 1947, 29, 15–32.
- Hasselbach, W.; Makinose, M. [The calcium pump of the “relaxing granules” of muscle and its dependence on ATP-splitting]. Biochem Z 1961, 333, 518–528.
- Hasselbach, W. Structural and enzymatic properties of the calcium transporting membranes of the sarcoplasmic reticulum. Ann. N. Y. Acad. Sci. 1966, 137, 1041–1048.
- Ebashi, S.; Lipmann, F. Adenosine triphosphate-linked concentration of calcium ions in a particulate fraction of rabbit muscle. J. Cell Biol. 1962, 14, 389–400.
- Weber, A.; Herz, R.; Reiss, I. On the Mechanism of the Relaxing Effect of Fragmented Sarcoplasmic Reticulum. J. Gen. Physiol. 1963, 46, 679–702.
- Jöbsis, F.; O’Connor, M. Calcium release and reabsorption in the sartorius muscle of the toad. Biochem. Biophys. Res. Commun. 1966, 25, 246–252.
- Slater, E.C.; Cleland, K.W. The effect of calcium on the respiratory and phosphorylative activities of heart-muscle sarcosomes. Biochem. J. 1953, 55, 566–580.
- Chance, B. On possible mechanisms for the control of electron transport in the respiratory chain. In Proceedings of the 3rd International Congress of Biochemistry, Brussels, Belgium, 1956.
- Stathopulos, P.B.; Zheng, L.; Li, G.-Y.; Plevin, M.J.; Ikura, M. Structural and Mechanistic Insights into STIM1-Mediated Initiation of Store-Operated Calcium Entry. Cell 2008, 135, 110–122.
- DeLuca, H.F.; Engstrom, G.W. CALCIUM UPTAKE BY RAT KIDNEY MITOCHONDRIA. Proc. Natl. Acad. Sci. USA 1961, 47, 1744–1750.
- Lehninger, A.L.; Rossi, C.S.; Greenawalt, J.W. Respiration-dependent accumulation of inorganic phosphate and Ca++ by rat liver mitochondria. Biochem. Biophys. Res. Commun. 1963, 10, 444–448.
- Mitchell, P.J. Coupling of Phosphorylation to Electron and Hydrogen Transfer by a Chemi-Osmotic type of Mechanism. Nat. Cell Biol. 1961, 191, 144–148.
- Denton, R.M.; Coore, H.G.; Martin, B.R.; Randle, P.J. Insulin activates Pyruvate Dehydrogenase in Rat Epididymal Adipose Tissue. Nat. New Biol. 1971, 231, 115–116.
- Ridgway, E.; Ashley, C. Calcium transients in single muscle fibers. Biochem. Biophys. Res. Commun. 1967, 29, 229–234.
- Rudolf, R.; Mongillo, M.; Rizzuto, R.; Pozzan, T. Looking forward to seeing calcium. Nat. Rev. Mol. Cell Biol. 2003, 4, 579–586.
- Takahashi, A.; Camacho, P.; Lechleiter, J.D.; Herman, B. Measurement of Intracellular Calcium. Physiol. Rev. 1999, 79, 1089–1125.
- Carafoli, E. The interplay of mitochondria with calcium: An historical appraisal. Cell Calcium 2012, 52, 1–8.
- Rizzuto, R.; Brini, M.; Pozzan, T. Targeting Recombinant Aequorin to Specific Intracellular Organelles. In Methods in Cell Biology; Elsevier B.V.: Amsterdam, The Netherlands, 1994; Volume 40, pp. 339–358.
- De Giorgi, F.; Brini, M.; Bastianutto, C.; Marsault, R.; Montero, M.; Pizzo, P.; Rossi, R.; Rizzuto, R. Targeting aequorin and green fluorescent protein to intracellular organelles. Gene 1996, 173, 113–117.
- Marsault, R.; Murgia, M.; Pozzan, T.; Rizzuto, R. Domains of high Ca2+ beneath the plasma membrane of living A7r5 cells. EMBO J. 1997, 16, 1575–1581.
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca2+ Responses. Science 1998, 280, 1763–1766.
- Pinton, P.; Pozzan, T.; Rizzuto, R. The Golgi apparatus is an inositol 1,4,5-trisphosphate-sensitive Ca2+ store, with functional properties distinct from those of the endoplasmic reticulum. EMBO J. 1998, 17, 5298–5308.
- Hayashi, T.; Rizzuto, R.; Hajnoczky, G.; Su, T.-P. MAM: more than just a housekeeper. Trends Cell Biol. 2009, 19, 81–88.
- Rizzuto, R.; Brini, M.; Murgia, M.; Pozzan, T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 1993, 262, 744–747.
- Perocchi, F.; Gohil, V.M.; Girgis, H.S.; Bao, X.R.; McCombs, J.E.; Palmer, A.E.; Mootha, V.K. MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nat. Cell Biol. 2010, 467, 291–296.
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabò, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340.
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nat. Cell Biol. 2011, 476, 341–345.
- Kovács-Bogdán, E.; Sancak, Y.; Kamer, K.J.; Plovanich, M.; Jambhekar, A.; Huber, R.J.; Myre, M.A.; Blower, M.D.; Mootha, V.K. Reconstitution of the mitochondrial calcium uniporter in yeast. Proc. Natl. Acad. Sci. USA 2014, 111, 8985–8990.
- Fan, M.; Zhang, J.; Tsai, C.-W.; Orlando, B.J.; Rodriguez, M.; Xu, Y.; Liao, M.; Tsai, M.-F.; Feng, L. Structure and mechanism of the mitochondrial Ca2+ uniporter holocomplex. Nat. Cell Biol. 2020, 582, 129–133.
- Wu, W.; Shen, Q.; Zhang, R.; Qiu, Z.; Wang, Y.; Zheng, J.; Jia, Z. The structure of the MICU 1- MICU 2 complex unveils the regulation of the mitochondrial calcium uniporter. EMBO J. 2020, 39.
- Wang, Y.; Han, Y.; She, J.; Nguyen, N.X.; Mootha, V.K.; Bai, X.-C.; Jiang, Y. Structural insights into the Ca2+-dependent gating of the human mitochondrial calcium uniporter. eLife 2020, 9, 1–19.
- Wang, C.; Jacewicz, A.; Delgado, B.D.; Baradaran, R.; Long, S.B. Structures reveal gatekeeping of the mitochondrial Ca2+ uniporter by MICU1-MICU2. eLife 2020, 9, 1–30.
- Nguyen, N.X.; Armache, J.-P.; Lee, C.; Yang, Y.; Zeng, W.; Mootha, V.K.; Cheng, Y.; Bai, X.-C.; Jiang, Y. Cryo-EM structure of a fungal mitochondrial calcium uniporter. Nat. Cell Biol. 2018, 559, 570–574.
- Yoo, J.; Wu, M.; Yin, Y.; Herzik, M.A., Jr.; Lander, G.C.; Lee, S.-Y. Cryo-EM structure of a mitochondrial calcium uniporter. Science 2018, 361, 506–511.
- Fan, C.; Fan, M.; Orlando, B.J.; Fastman, N.M.; Zhang, J.; Xu, Y.; Chambers, M.G.; Xu, X.; Perry, K.; Liao, M.; et al. X-ray and cryo-EM structures of the mitochondrial calcium uniporter. Nat. Cell Biol. 2018, 559, 575–579.
- Baradaran, R.; Wang, C.; Siliciano, A.F.; Long, S.B. Cryo-EM structures of fungal and metazoan mitochondrial calcium uniporters. Nat. Cell Biol. 2018, 559, 580–584.
- Oxenoid, K.; Dong, Y.; Cao, C.; Cui, T.; Sancak, Y.; Markhard, A.L.; Grabarek, Z.; Kong, L.; Liu, Z.; Ouyang, B.; et al. Architecture of the mitochondrial calcium uniporter. Nat. Cell Biol. 2016, 533, 269–273.
- Wang, Y.; Nguyen, N.X.; She, J.; Zeng, W.; Yang, Y.; Bai, X.-C.; Jiang, Y. Structural Mechanism of EMRE-Dependent Gating of the Human Mitochondrial Calcium Uniporter. Cell 2019, 177, 1252–1261.
- Zhuo, W.; Zhou, H.; Guo, R.; Yi, J.; Zhang, L.; Yu, L.; Sui, Y.; Zeng, W.; Wang, P.; Yang, M. Structure of intact human MCU supercomplex with the auxiliary MICU subunits. Protein Cell 2021, 12, 220–229.
- Pan, X.; Liu, J.; Nguyen, T.; Liu, C.; Sun, J.; Teng, Y.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 2013, 15, 1464–1472.
- Kwong, J.Q.; Lu, X.; Correll, R.N.; Schwanekamp, J.A.; Vagnozzi, R.J.; Sargent, M.A.; York, A.J.; Zhang, J.; Bers, D.M.; Molkentin, J.D. The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart. Cell Rep. 2015, 12, 15–22.
- Luongo, T.S.; Lambert, J.P.; Yuan, A.; Zhang, X.; Gross, P.; Song, J.; Shanmughapriya, S.; Gao, E.; Jain, M.; Houser, S.R.; et al. The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep. 2015, 12, 23–34.
- Drago, I.; Davis, R.L. Inhibiting the Mitochondrial Calcium Uniporter during Development Impairs Memory in Adult Drosophila. Cell Rep. 2016, 16, 2763–2776.
- Choi, S.; Quan, X.; Bang, S.; Yoo, H.; Kim, J.; Park, J.; Park, K.-S.; Chung, J. Mitochondrial calcium uniporter in Drosophila transfers calcium between the endoplasmic reticulum and mitochondria in oxidative stress-induced cell death. J. Biol. Chem. 2017, 292, 14473–14485.
- Hamilton, J.; Brustovetsky, T.; Rysted, J.E.; Lin, Z.; Usachev, Y.M.; Brustovetsky, N. Deletion of mitochondrial calcium uniporter incompletely inhibits calcium uptake and induction of the permeability transition pore in brain mitochondria. J. Biol. Chem. 2018, 293, 15652–15663.
- Gherardi, G.; Nogara, L.; Ciciliot, S.; Fadini, G.P.; Blaauw, B.; Braghetta, P.; Bonaldo, P.; De Stefani, D.; Rizzuto, R.; Mammucari, C. Loss of mitochondrial calcium uniporter rewires skeletal muscle metabolism and substrate preference. Cell Death Differ. 2019, 26, 362–381.
- Kwong, J.Q.; Huo, J.; Bround, M.J.; Boyer, J.G.; Schwanekamp, J.A.; Ghazal, N.; Maxwell, J.T.; Jang, Y.C.; Khuchua, Z.; Shi, K.; et al. The mitochondrial calcium uniporter underlies metabolic fuel preference in skeletal muscle. JCI Insight 2018, 3.
- Huang, G.; Vercesi, A.E.; Docampo, R. Essential regulation of cell bioenergetics in Trypanosoma brucei by the mitochondrial calcium uniporter. Nat. Commun. 2013, 4, 1–14.
- Prudent, J.; Popgeorgiev, N.; Bonneau, B.; Thibaut, J.; Gadet, R.; Lopez, J.; Gonzalo, P.; Rimokh, R.; Manon, S.; Houart, C.; et al. Bcl-wav and the mitochondrial calcium uniporter drive gastrula morphogenesis in zebrafish. Nat. Commun. 2013, 4, 2330.
- Tomar, D.; Jaña, F.; Dong, Z.; Quinn, W.J.; Jadiya, P.; Breves, S.L.; Daw, C.C.; Srikantan, S.; Shanmughapriya, S.; Nemani, N.; et al. Blockade of MCU-Mediated Ca2+ Uptake Perturbs Lipid Metabolism via PP4-Dependent AMPK Dephosphorylation. Cell Rep. 2019, 26, 3709–3725.
- Soman, S.; Keatinge, M.; Moein, M.; Da Costa, M.; Mortiboys, H.; Skupin, A.; Sugunan, S.; Bazala, M.; Kuznicki, J.; Bandmann, O. Inhibition of the mitochondrial calcium uniporter rescues dopaminergic neurons inpink1−/−zebrafish. Eur. J. Neurosci. 2017, 45, 528–535.
- Murphy, E.; Pan, X.; Nguyen, T.; Liu, J.; Holmström, K.M.; Finkel, T. Unresolved questions from the analysis of mice lacking MCU expression. Biochem. Biophys. Res. Commun. 2014, 449, 384–385.
- Álvarez-Illera, P.; García-Casas, P.; Fonteriz, R.I.; Montero, M.; Alvarez, J. Mitochondrial Ca2+ Dynamics in MCU Knockout C. elegans Worms. Int. J. Mol. Sci. 2020, 21, 8622.
- Raffaello, A.; De Stefani, D.; Sabbadin, D.; Teardo, E.; Merli, G.; Picard, A.; Checchetto, V.; Moro, S.; Szabò, I.; Rizzuto, R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013, 32, 2362–2376.
- Chiurillo, M.A.; Lander, N.; Bertolini, M.S.; Storey, M.; Vercesi, A.E.; Docampo, R. Different Roles of Mitochondrial Calcium Uniporter Complex Subunits in Growth and Infectivity of Trypanosoma cruzi. mBio 2017, 8, e00574-17.
- Fieni, F.; Lee, S.B.; Jan, Y.N.; Kirichok, Y. Activity of the mitochondrial calcium uniporter varies greatly between tissues. Nat. Commun. 2012, 3, 1–12.
- Huo, J.; Lu, S.; Kwong, J.Q.; Bround, M.J.; Grimes, K.M.; Sargent, M.A.; Brown, M.E.; Davis, M.E.; Bers, D.M.; Molkentin, J.D. MCUb Induction Protects the Heart From Postischemic Remodeling. Circ. Res. 2020, 127, 379–390.
- Lambert, J.P.; Luongo, T.S.; Tomar, D.; Jadiya, P.; Gao, E.; Zhang, X.; Lucchese, A.M.; Kolmetzky, D.W.; Shah, N.S.; Elrod, J.W. MCUB Regulates the Molecular Composition of the Mitochondrial Calcium Uniporter Channel to Limit Mitochondrial Calcium Overload During Stress. Circulation 2019, 140, 1720–1733.
- Mammucari, C.; Raffaello, A.; Reane, D.V.; Gherardi, G.; De Mario, A.; Rizzuto, R. Mitochondrial calcium uptake in organ physiology: from molecular mechanism to animal models. Pflügers Arch. Eur. J. Physiol. 2018, 470, 1165–1179.
- Csordás, G.; Golenár, T.; Seifert, E.L.; Kamer, K.J.; Sancak, Y.; Perocchi, F.; Moffat, C.; Weaver, D.; Perez, S.D.L.F.; Bogorad, R.; et al. MICU1 Controls Both the Threshold and Cooperative Activation of the Mitochondrial Ca2+ Uniporter. Cell Metab. 2013, 17, 976–987.
- Mallilankaraman, K.; Doonan, P.; Cárdenas, C.; Chandramoorthy, H.C.; Müller, M.; Miller, R.; Hoffman, N.E.; Gandhirajan, R.K.; Molgó, J.; Birnbaum, M.J.; et al. MICU1 Is an Essential Gatekeeper for MCU-Mediated Mitochondrial Ca2+ Uptake that Regulates Cell Survival. Cell 2012, 151, 630–644.
- Liu, J.C.; Liu, J.; Holmström, K.M.; Menazza, S.; Parks, R.J.; Fergusson, M.M.; Yu, Z.-X.; Springer, D.A.; Halsey, C.; Liu, C.; et al. MICU1 Serves as a Molecular Gatekeeper to Prevent In Vivo Mitochondrial Calcium Overload. Cell Rep. 2016, 16, 1561–1573.
- Logan, C.V.; Szabadkai, G.; A Sharpe, J.; A Parry, D.; Torelli, S.; Childs, A.-M.; Kriek, M.; Phadke, R.; A Johnson, C.; Roberts, N.Y.; et al. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat. Genet. 2013, 46, 188–193.
- Antony, A.N.; Paillard, M.; Moffat, C.; Juskeviciute, E.; Correnti, J.; Bolon, B.; Rubin, E.; Csordás, G.; Seifert, E.L.; Hoek, J.B.; et al. MICU1 regulation of mitochondrial Ca2+ uptake dictates survival and tissue regeneration. Nat. Commun. 2016, 7, 10955.
- Nemani, N.; Dong, Z.; Daw, C.C.; Madaris, T.R.; Ramachandran, K.; Enslow, B.T.; Rubannelsonkumar, C.S.; Shanmughapriya, S.; Mallireddigari, V.; Maity, S.; et al. Mitochondrial pyruvate and fatty acid flux modulate MICU1-dependent control of MCU activity. Sci. Signal. 2020, 13, eaaz6206.
- Gottschalk, B.; Klec, C.; Leitinger, G.; Bernhart, E.; Rost, R.; Bischof, H.; Madreiter-Sokolowski, C.T.; Radulović, S.; Eroglu, E.; Sattler, W.; et al. MICU1 controls cristae junction and spatially anchors mitochondrial Ca2+ uniporter complex. Nat. Commun. 2019, 10, 1–17.
- Kamer, K.J.; Sancak, Y.; Fomina, Y.; Meisel, J.D.; Chaudhuri, D.; Grabarek, Z.; Mootha, V.K. MICU1 imparts the mitochondrial uniporter with the ability to discriminate between Ca2+ and Mn2+. Proc. Natl. Acad. Sci. USA 2018, 115, E7960–E7969.
- Reane, D.V.; Vallese, F.; Checchetto, V.; Acquasaliente, L.; Butera, G.; De Filippis, V.; Szabò, I.; Zanotti, G.; Rizzuto, R.; Raffaello, A. A MICU1 Splice Variant Confers High Sensitivity to the Mitochondrial Ca 2+ Uptake Machinery of Skeletal Muscle. Mol. Cell 2016, 64, 760–773.
- Plovanich, M.; Bogorad, R.L.; Sancak, Y.; Kamer, K.J.; Strittmatter, L.; Li, A.A.; Girgis, H.S.; Kuchimanchi, S.; De Groot, J.; Speciner, L.; et al. MICU2, a Paralog of MICU1, Resides within the Mitochondrial Uniporter Complex to Regulate Calcium Handling. PLoS ONE 2013, 8, e55785.
- Wu, W.; Shen, Q.; Lei, Z.; Qiu, Z.; Li, D.; Pei, H.; Zheng, J.; Jia, Z. The crystal structure of MICU 2 provides insight into Ca 2+ binding and MICU 1- MICU 2 heterodimer formation. EMBO Rep. 2019, 20, e47488.
- Payne, R.; Hoff, H.; Roskowski, A.; Foskett, J.K. MICU2 Restricts Spatial Crosstalk between InsP 3 R and MCU Channels by Regulating Threshold and Gain of MICU1-Mediated Inhibition and Activation of MCU. Cell Rep. 2017, 21, 3141–3154.
- Bick, A.G.; Wakimoto, H.; Kamer, K.J.; Sancak, Y.; Goldberger, O.; Axelsson, A.; DeLaughter, D.M.; Gorham, J.M.; Mootha, V.K.; Seidman, J.G.; et al. Cardiovascular homeostasis dependence on MICU2, a regulatory subunit of the mitochondrial calcium uniporter. Proc. Natl. Acad. Sci. USA 2017, 114, E9096–E9104.
- Patron, M.; Granatiero, V.; Espino, J.; Rizzuto, R.; De Stefani, D. MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ. 2018, 26, 179–195.
- Márkus, N.M.; Hasel, P.; Qiu, J.; Bell, K.F.S.; Heron, S.; Kind, P.C.; Dando, O.; Simpson, T.I.; Hardingham, G.E. Expression of mRNA Encoding Mcu and Other Mitochondrial Calcium Regulatory Genes Depends on Cell Type, Neuronal Subtype, and Ca2+ Signaling. PLoS ONE 2016, 11, e0148164.
- Xing, Y.; Wang, M.; Wang, J.; Nie, Z.; Wu, G.; Yang, X.; Shen, Y. Dimerization of MICU Proteins Controls Ca2+ Influx through the Mitochondrial Ca2+ Uniporter. Cell Rep. 2019, 26, 1203–1212.
- Sancak, Y.; Markhard, A.L.; Kitami, T.; Kovács-Bogdán, E.; Kamer, K.J.; Udeshi, N.D.; Carr, S.A.; Chaudhuri, D.; Clapham, D.E.; Li, A.; et al. EMRE Is an Essential Component of the Mitochondrial Calcium Uniporter Complex. Science 2013, 342, 1379–1382.
- Yamamoto, T.; Yamagoshi, R.; Harada, K.; Kawano, M.; Minami, N.; Ido, Y.; Kuwahara, K.; Fujita, A.; Ozono, M.; Watanabe, A.; et al. Analysis of the structure and function of EMRE in a yeast expression system. Biochim. Biophys. Acta (BBA) Bioenerg. 2016, 1857, 831–839.
- Vais, H.; Mallilankaraman, K.; Mak, D.-O.D.; Hoff, H.; Payne, R.; Tanis, J.E.; Foskett, J.K. EMRE Is a Matrix Ca 2+ Sensor that Governs Gatekeeping of the Mitochondrial Ca 2+ Uniporter. Cell Rep. 2016, 14, 403–410.
- Tsai, M.-F.; Phillips, C.B.; Ranaghan, M.; Tsai, C.-W.; Wu, Y.; Willliams, C.; Miller, C. Dual functions of a small regulatory subunit in the mitochondrial calcium uniporter complex. eLife 2016, 5.
- König, T.; Tröder, S.E.; Bakka, K.; Korwitz, A.; Richter-Dennerlein, R.; Lampe, P.A.; Patron, M.; Mühlmeister, M.; Guerrero-Castillo, S.; Brandt, U.; et al. The m -AAA Protease Associated with Neurodegeneration Limits MCU Activity in Mitochondria. Mol. Cell 2016, 64, 148–162.

