The notion of the active role of mitochondria in the decoding and shaping of intracellular Ca2+ signals dates back at the end of the 19th century. However, the identity of the molecule(s) involved in Ca2+ ion transport into mitochondria remained elusive for decades. Only in the last ten years, the factors, and the relative coding genes, mediating Ca2+ entry in mitochondria started to be genetically and biochemically described. The gene for the pore-forming unit of the mitochondrial Ca2+ channel was discovered in 2011, and its product was named mitochondrial Ca2+ uniporter or MCU. The mitochondria Ca2+ uptake regulator 1 gene, MICU1, was cloned one year before, in 2010. The increasing interest of the scientific community towards mitochondrial Ca2+ signaling and metabolism in the subsequent years led to the identification of many other MCU components and to the description of their 3D structure and physiological role. Here, we will present a brief overview of the land marking discoveries in the history of mitochondrial Ca2+ studies.
- MCU
- mitochondrial Ca2+ uniporter
- Ca2+ signaling
- mitochondrial metabolism
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1. Introduction
Ca
2+.ions are involved in the decoding of a vast range of stimuli [1–3] and mitochondria play a fundamental role in the orchestration of cellular Ca
2+ ions participate in the decoding of a vast range of stimuli and the variety of cellular components involved in the Casignals. Indeed, mitochondria efficiently uptake Ca
2+ signal transduction is extremely wide, including basically all kinds of components, organelles, and molecules [1][2][3]. The research studies on Caupon Ca
2+ second messenger started more than one hundred years ago, with the initial recognition of the role of Carelease from ER or extracellular Ca
2+ in muscle cell contraction [4]. Since then, the understanding of Caentry [4] thus buffering the cytosolic Ca
2+ signaling regulation and dynamics has progressively increased leading to the definition of the concept of Caraise and tuning global Ca
2+ compartmentalization and to the demonstration of the existence of microdomains of local high Casignaling. Moreover, the increase of Ca
2+ concentration [5], which are crucial for the fine-tuning and correct triggering of the Cain the mitochondrial matrix stimulates both Ca
2+-dependent cellular effects [1].-sensitive mitochondrial dehydrogenases [5–7] and respiratory chain complexes [8,9] thus boosting cellular oxidative metabolism.
We provide here a brief overview of the milestone achievements in the history of mitochondrial Ca
2+ signals. Indeed, mitochondria are not a store of rapidly releasable Caresearch, with a particular focus on the recent findings on the mitochondrial Ca
2+ (such as the ER), but rather they efficiently accumulate Ca2+ upon Ca2+ entry from the extracellular space or upon release from ER Ca2+ stores [6]. Upon cytosolic Ca2+ elevation, the entry of Ca2+ into mitochondria exerts a central function in the modulation of cell metabolism. Mitochondria host the enzymes and complexes of the TCA cycle, fatty acid oxidation (FAO), and oxidative phosphorylation (OXPHOS) thus representing the site of the major metabolic pathways and enzymes for cell energy supply, which deserved them the name of ‘cellular powerhouses’. Interestingly, Ca2+ entry and oxidative activity are two strictly intertwined aspects of mitochondrial physiology. The increase of the mitochondrial matrix Ca2+ level stimulates both Ca2+-sensitive dehydrogenases [7][8][9] and respiratory chain complexes [10][11] resident in the organelles, fueling the TCA cycle activity as well as aerobic respiration and thus boosting the overall oxidative metabolism. This makes mitochondria the central hubs for the rapid and effective adaptation of cell metabolism to the changes in energy requirements that are typically decoded as variations of intracellular Ca2+ concentration.uniporter complex and its regulators.
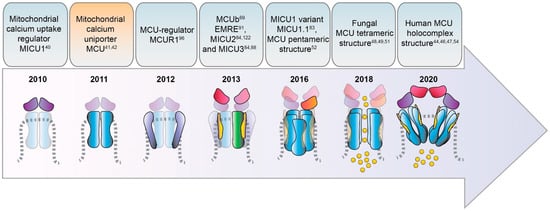
Timeline of the identification of MCU complex components.
2. Mitochondrial Ca2+ Tsimeline of MCU Identificgnaling in the pre-MCU eration
The first report about the relevance of Ca
2+ ions being relevant to organ physiology dates back to more than one century ago when the first report on the physiological action of Caions to organ physiology dates back to the work of Ringer in 1883 [10], who described the capacity of Ca
2+ ions appeared in 1883 [4]. At that time, Ringer described the effects of Cato induce organ contraction when added to isolated frog hearts. After that, the cardiac physiologists Slater and Cleland identified some subcellular compartments able to accumulate Ca
2+ addition to isolated frog hearts and demonstrated that the supplementation of Cathat they called "sarcosomes" [11]. Interestingly, these “sarcosomes” were indeed isolated mitochondria, to which the addition of Ca
2+ in the perfusion solution actively induces and sustains the contraction of the organ ex vivo [4]. This seminal observation revealed that Cacaused the block of their oxidative phosphorylation activity. Thus, Ca
2+ is a fundamental messenger within cells, a concept that then extended to virtually every cell type and physiological and pathological process, giving rise to a broad field of study commonly referred to as the field of intracellular Caions behaved as mitochondria uncoupler.
It was in the early 60′s, even before the postulation of the chemiosmotic theory by Peter Mitchell [12] that the concept of Ca
2+ signaling. The intrinsic ability of contracting myocytes to operate ex vivo and to rapidly and effectively respond to environmental condition changes made them the ideal experimental system for the investigation of the role of Cabeing actively taken up by energized mitochondria was experimentally validated [13–15].
Soon after that, the group of Denton discovered that three critical oxidative enzymes resident in mitochondria (pyruvate dehydrogenase phosphatase, NAD-isocitrate dehydrogenase and oxoglutarate dehydrogenase) are subjected to a Ca
2+ in organ and cell physiology and was extensively exploited by researchers in the following years.-dependent modulation [5–7].
Despite all these conceptual advancements, two major issues continued to puzzle the scientific community for a long time: i) the apparent paradox between the physiological low Ca
2+ stores to accumulate the cation required to sustain muscle contraction has been later postulated and demonstrated in 1947 by Heilbrunn [16]. However, although surprising, the identification of the sarco/endoplasmic reticulum (SR/ER) as the principal cellular Calevels (in the submicromolar range) present in the cytosol, [16–18], and the reduced Ca
2+ store came only 20 years later. It was in the 1960′s, with the identification of Caaffinity of the mitochondrial Ca
2+ pumping machinery on intracellular membranes (in particular the calcium pump of the sarcoplasmic reticulum, better known as SERCA) by three independent scientists [17][18][19][20] and the advent of new methodologies for the measurement of intracellular Cauptake machinery (in the order of several µM [19]); ii) the fundamental question about the molecular identity of the mitochondrial Ca
2+ concentration [21] that the ER and its specialized counterpart in muscle cells (the SR) were recognized the main cellular reservoir of Cachannel mediating Ca
2+.entry into mitochondria.
It took several decades to answer to those two enigmas. Indeed, thanks to the development of new genetic Ca
2+ [22]. Interestingly, these “sarcosomes” did not consist of ER but, instead, they corresponded to isolated mitochondria, to which the addition of Caprobes and recombinant fluorescent proteins specifically targeted to intracellular domains [20,21], it was possible to measure variations of Ca
2+ caused the block of their oxidative phosphorylation activity. Thus, Caconcentration in defined and limited areas of the cell. This led to the visualization of organelle dynamics and interorganelle contacts (as those between mitochondria and ER) and to the identification of microdomains of high Ca
2+ ions behaved as mitochondria uncouplers. Despite that, Caconcentration right at the mouth of the channel pores, exactly where mitochondria are located. This allows mitochondria to perceive a local cation concentration sufficient to meet the low affinity of the mitochondrial Ca
2+ appeared to exert a peculiar inhibitory action on mitochondrial OXPHOS activity, which differed from the other irreversible uncouplers known at that period (dinitrophenol, dicoumarol, rotenone, antimycin A, azide, or cyanide), due to the reversibility of its action [23]. This assigned to Cauptake machinery [21,22]. The answer to the second fundamental question of the mitochondrial signaling field came with the discovery of the molecular identity of the mitochondrial Ca
2+ ions a functional role in mitochondria activity.uniporter (MCU) and its multiple regulators.
3. DiscovIdery and Characterizntification of the MCU Complex Components
3.1. MCU
The discovery of MCU is due to two pioneering studies published in 2011 [23,24] (Figure 1) that identified the MCU pore-forming protein and cloned its coding gene CCDC109A. MCU is a highly conserved 40 kDa polypeptide ubiquitously expressed in plants, metazoans, protozoans, and fungi but absent in yeast [25], it oligomerizes to form the active Ca
2+ that follows the IP3-generating agonist in stimulated cells. Of note, the blunted mitochondrial Cachannel within the inner mitochondrial membrane (IMM) and directly interacts with the mitochondrial Ca
2+ uptake response occurs without changes in the mitochondria morphology or membrane potential of the MCU-silenced cells [41]. On the contrary, MCU overexpression enhances agonist-induced mitochondrial Cauptake regulator 1 protein MICU1 [26].
Recently, the structure of the human MCU together with its auxiliary component EMRE was obtained [27]. Each human MCU arranges in tetramers and each subunit complexes with one EMRE peptide. The silencing of MCU blunted the mitochondrial Ca
2+ uptake in mammalian cells. In addition, in vitro experiments, in which recombinant MCU proteins were inserted in a planar lipid bilayer, showed that this pore-subunit alone is sufficient to form the channel. Indeed, in this setting, the MCU electrophysiological activity is completely abolished by the addition of the known inhibitor Ruthenium Red, firmly pointing at MCU as the genuine core component of the mitochondrial Cauptake response in HeLa cells [23]. On the contrary, MCU overexpression enhances agonist-induced mitochondrial Ca
2+ machinery.uptake in mammalian cells. In addition, the precise description of the molecular interactions between MCU and its regulators EMRE-MICU1-MICU2 in the complex (called holocomplex or uniplex) has been also obtained both in the presence and absence of Ca
2+ transport since, if substituted with uncharged residues, MCU mutants failed to rescue the mitochondrial Ca2+ uptake in MCU-silenced cells [41][42]. However, the definitive description of MCU 3D structure had to wait till the very last years, when cryo-EM and X-ray diffraction analyses finally allowed the resolution of full-length MCU structure [44][45][46][47][48][49][50][51]. These studies coherently confirmed that purified MCU from different sources (fungi and metazoan) arranges in a tetramer, confuting previous assumptions on a putative pentameric MCU architecture [52]. Notably, the cryo-EM data also unveiled the exact position of the MCU channel selectivity filter, in which the DIME motif is fundamental for the coordination of Ca2+ ions and which was definitively shown to reside at the beginning of the second transmembrane α-helix [50] and not in the linker region between the two transmembrane helices, as previously suggested [52].[28] (Figure 2).
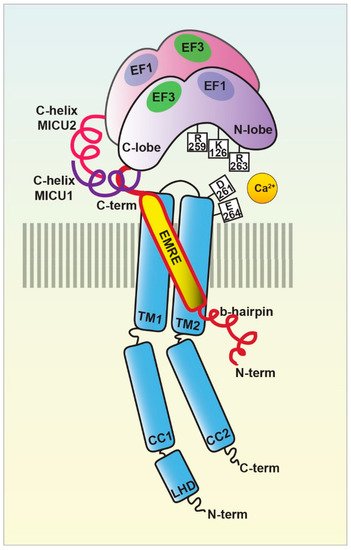
Figure 2. The MCU holocomplex structure. Schematic representation of the MCU holocomplex (uniplex) components and their relevant domains: the pore-forming subunit MCU (light blue) with the two transmembrane (TM) and coiled-coil (CC) domains and the linker helix domain (LHD); the essential mitochondrial Ca
uniporter regulator EMRE (yellow); the mitochondrial Ca
uptake proteins MICU1 (violet) and MICU2 (purple), with the EF-hands relevant for the MICU dimer interaction highlighted. The critical residues of the MCU DIME motif forming the Ca
selectivity filter are indicated, together with the MICU1 residues of the K-R ring coordinating the MCU acidic region.
In the presence of the ion, the multiprotein holocomplex shows a two-fold symmetry and consists of two V-shaped MCU-EMRE heterotetramers and two MICU1-MICU2 heterodimers that bridge the tops of the MCU-EMRE tetramers (Figure 1) through the interaction between MICU1 and EMRE [29] (Figure 2). Differently, in the absence of Ca
2+-free and Ca, the holocomplex adopts a less stable conformation, where the MICU1-MICU2 heterodimer blocks the channel pore formed by the MCU transmembrane domains [29] (Figures 2 and 3).
Sequence and topology analyses revealed that both the N- and C-termini of MCU are located in the mitochondrial matrix and that MCU is endowed with two transmembrane domains, linked by a short highly conserved acidic loop exposed in the intermembrane space (IMS) which contains the so-called acidic “DIME” motif. The acidic residues present in this stretch are critical for the Ca
2+-bound state by several independent reports. The gating mechanism by which MICU1 regulates the uniporter activity via the conformational change triggered by Cacoordination and constitute tha selectivity filter of the MCU channel [23,24].
More recently, the structure of the human MCU together with its auxiliary component EMRE was obtained [27]. Each human MCU arranges in tetramers and each subunit complexes with one EMRE peptide. Human MCU appears organized in various domains, which are: i) the N-terminal domain (NTD), ii) the linker helix domain (LHD) —absent in fungi—, iii) the coiled-coil domains (CC), and iv) the transmembrane domains (TM) (Figure 2).
The gating mechanism by which MICU1 regulates the uniporter activity via the conformational change triggered by Ca
2+ was finally unveiled [44]. Furthermore, the precise description of the molecular interactions between MCU-EMRE-MICU1-MICU2 in the human MCU supercomplex (MEMMS) has been also obtained [54] (Figure 2). Indeed, MEMMS appears as a 480 kDa integral unit where EMRE coordinates the matrix gate of the MCU channel and MICU proteins interact with the C-terminus of EMRE in the IMS thus enhancing Cawas finally unveiled in 2020 [30]. Indeed, EMRE coordinates the matrix gate of the MCU channel and interacts with MICU proteins in the IMS enhancing Ca
2+influx through the MCU pore in high [Ca
2+] conditions [54] (Figure 3). Finally, the distinct Ca] conditions [28] (Figure 2 and 3). In the presence of Ca
2+-dependent assembly conformations of the beetle and human MCU holocomplexes with human MICUs have also been detailed [46][47]. In the presence of Ca, the multiprotein complex shows a two-fold symmetry and consists of two V-shaped MCU-EMRE tetrameric subcomplexes and two MICU1-MICU2 heterodimers that bridge the tops of the subcomplexes (Figure 1). In this setting, the assembly of the MICU1-MICU2 heterodimers to the MCU-EMRE subcomplexes is ensured by the interaction between MICU1 and EMRE [29] (Figure 2). Differently, in the absence of Ca
2+, the multiprotein complex shows a two-fold symmetry and consists of two V-shaped MCU-EMRE tetrameric subcomplexes and two MICU1-MICU2 heterodimers that bridge the tops of the subcomplexes (Figure 1). In this setting, the assembly of the MICU1-MICU2 heterodimers to the MCU-EMRE subcomplexes is ensured by the interaction between MICU1 and EMRE [47] (Figure 2). Differently, in the absence of Ca2+, the holocomplex adopts alternative less stable conformations with both monomeric and dimeric forms of the MCU-EMRE tetramers, where the MICU1-MICU2 heterodimer block the channel entrance formed by MCU transmembrane domains [47] (Figure 2 and Figure 3)., the holocomplex adopts alternative less stable conformations with both monomeric and dimeric forms of the MCU-EMRE tetramers, where the MICU1-MICU2 heterodimer block the channel entrance formed by MCU transmembrane domains [29] (Figures 2 and 3).
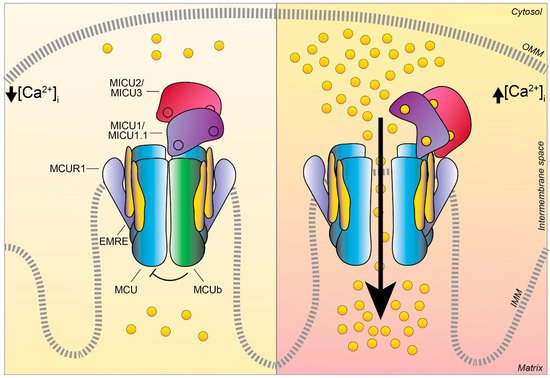
The MCU complex activity at low and high intracellular Ca
concentration. Schematic representation of the proteins involved in the MCU complex-mediated mitochondrial Ca
uptake: the pore-forming subunits MCU (light blue) and MCUb (green), the essential mitochondrial Ca
uniporter regulator EMRE (yellow), the mitochondrial Ca
uptake proteins MICU1 / MICU1.1 (violet), MICU2/ MICU3 (purple), and the MCU regulator 1 MCUR1 (light violet). The EF-hand Ca
binding domains of MICU proteins are indicated as little circles. At low intracellular Ca
concentration, the cation does not permeate through the MCU channel since the heterodimers formed by MICU1/MICU1.1–MICU2/MICU3 block the channel pore, thus preventing Ca
flux in resting conditions. Differently, at high intracellular Ca
concentration, MICU proteins undergo conformational changes relieving the inhibition on MCU and positively regulating channel activity, leading to an efficient mitochondrial Ca
uptake. OMM, outer mitochondrial membrane; IMM, inner mitochondrial membrane.
In 2013, three independent groups showed that the MCU silencing/knockout or its overexpression affect survival in different in vivo models [31–33]. The MCU
–/– mouse [55] develops normally and displays minor defects without signs of impaired cell survival. Under stress conditions, relatively mild metabolic alterations were observed, such as increased plasma lactate levels in line with impaired exercise performance. However, the same group soon after those findings, showed that embryos from MCUmouse [31] develops normally and displays minor defects (only mild metabolic alterations after stress conditions) without signs of impaired cell survival. However, the same group showed that, in a pure C57BL/6 background, MCU
−/− mice in a pure C57BL/6 background were not viable, displaying embryonic lethality at around E 11.5–E 13.5, thus suggesting a major involvement of mitochondrial Camouse embryos were not viable, displaying lethality at around E 11.5–E 13.5, thus suggesting a major involvement of mitochondrial Ca
2+ uptake in organ metabolism and organism development that was compensated in a mixed genetic background [67].uptake in organ metabolism and organism development that was compensated in a mixed genetic background [34].
Interestingly, some years later, it has been reported that the block of MCU-dependent Ca
2+ uptake affects Drosophila melanogaster development. In particular, the inhibition of the uniporter activity has been shown to be detrimental for memory establishment during the papulation stage. Indeed, during the development of adult flies, alterations in the structural and functional neuronal substrates, crucial for memory formation, occur in the MCU deficient fly [58].uptake affects Drosophila melanogaster development and memory establishment [35].
3.2. MCUb
MCU is not the only pore-forming subunit of the mitochondrial Ca
2+ uniporter, since an alternative MCU isoform exists, named MCUb, which crosses the IMM and associates to MCU to form the calcium channel [69] (Figure 1 and Figure 3). The MCUb protein is encoded by the MCU CCDC109a paralog gene CCDC109b. Interestingly, this gene is found in vertebrates, but it is not present in other organisms in which MCU is expressed, such as plants, Nematoda, and Arthropoda. Despite the high structural similarity with MCU, MCUb sequence presents two critical aminoacidic substitutions in the loop region and in the TM1 domain, which explains its inability to transport Cauniporter, since an alternative MCU isoform was discovered in 2013 and named MCUb. It crosses the IMM and associates to MCU to form the calcium channel [36] (Figures 1 and 3). Despite the high similarity with MCU, MCUb presents differences in two critical aminoacidic positions in the loop region and in the TM1 domain, which impede Ca
2+. Indeed, MCUb acts as a negative regulator of MCU activity, drastically reducing mitochondrial Catransport. Thus, MCUb acts as a MCU negative regulator, drastically reducing mitochondrial Ca
2+ currents in vitro in planar lipid bilayer experiments and also when overexpressed in mammalian cells [69]. On the contrary, in other organisms, such as trypanosomatid species, the ortholog of MCUb is capable to conduct the cation and its overexpression facilitates mitochondrial Cacurrents both in vitro and in mammalian cells [36]. However, in other organisms, such as trypanosomatid species, the orthologue of MCUb is capable to conduct the cation and its overexpression facilitates mitochondrial Ca
2+ uptake [70].uptake [37].
Notably, MCUb is expressed at different levels in different tissues, and the MCUb:MCU proportion results a distinctive feature of each cell-type ensuring the appropriate mitochondrial Ca
2+ current to each cell type [69][71]. For instance, a high MCUb/MCU ratio (3:1) is typical of cells with low mitochondrial Cacurrent to each of them [36,38]. As an example, adult cardiomyocytes have a high MCUb/MCU ratio (3:1) and display relatively modest mitochondrial Ca
2+ transients, such as adult cardiomyocytes (Figure 4). In fact, MCUb can be described as a protective gene in cardiac myocytes since i) its expression is transiently induced after ischemia-reperfusion injury and ii) transgenic mice overexpressing MCUb have a reduced mitochondrial Catransients (Figure 4). On the contray, skeletal muscle and neuronal cells have a low MCUb/MCU ratio (1:40) and are characterized by an extremely high capacity to accumulate Ca
2+ uptake ability, thus preventing Ca2+ overload, which is sufficient to protect myocytes from ischemia-reperfusion injury and to decelerate their ongoing necrosis [72][73]. A low MCUb/MCU ratio (1:40) is instead a characteristic of tissues with an extremely high capacity of mitochondrial Ca2+ accumulation, such as skeletal muscle [69][74] (Figure 4).in mitochondria [36,39] (Figure 4).
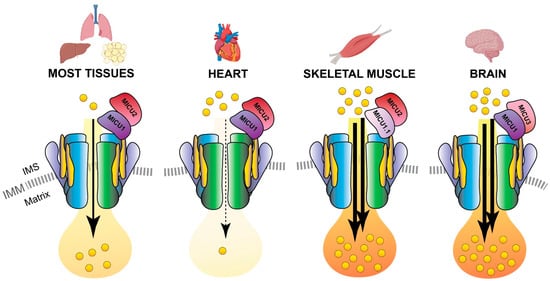
MCU holocomplex composition in different tissues. Schematic representation of the tissue-specific components of the MCU holo-complex. The presence of a relatively high MCUb:MCU ratio in the heart ensures a re-duced Ca
load in cardiomyocyte mitochondria. On the contrary, the expression of the MICU1.1 variant and MICU3 determines an elevated Ca
flux in the mitochondria of skeletal muscle fibers and neurons, respectively. IMS, inter-membrane space; IMM, inner mitochondrial membrane.
3.3. MICU1
MICU1 was actually the first component to be described as the regulator of the long-sought MCU channel and its identification even anticipated that of the pore-forming subunit MCU (Figure 1). Indeed, in 2010, an integrative comparative strategy revealed the product of the CBARA gene to be the mitochondrial calcium uptake 1 (MICU1). MICU1 resides in the IMS and does not take part to the pore-forming domain of the channel, but regulates its activity in a Ca
2+-dependent way [40][75]. Indeed, after the N-terminal mitochondrial targeting sequence, MICU1 shows two canonical EF-hand Ca-dependent way [26,40]since it possesses two canonical Ca
2+ binding domains that confers the Cabinding EF-hand domains that confers the Ca
2+-sensitivity (Figure 2 and Figure 3). In the last decade, several lines of evidence confirmed the initial hypothesis about the role of MICU1 as both MCU gatekeeper at low Ca-sensitivity (Figures 2 and 3). This allows MICU1 to act as both MCU gatekeeper, at low Ca
2+ concentration and MCU positive regulator at high Caconcentration, and MCU positive regulator, at high Ca
2+ concentration, thus explaining the sigmoid cooperative effect of the MCU activation curve [75].concentration [40].
The downregulation of MICU1 abolishes mitochondrial Ca
2+ influx in intact and permeabilized HeLa cells [40]. Later studies also demonstrated that the absence of MICU1 also leads to an adaptive Cainflux in cells [26] and leads to an adaptive Ca
2+ accumulation inside the mitochondria matrix, triggering excessive ROS production and the consequent higher sensitivity to apoptotic stress. In addition, the ability of MICU1 to sense cytosolic Ca2+ levels confers to MICU1 the capacity to set the threshold for the activation of mitochondrial Ca2+ uptake. Nevertheless, this occurs without altering the overall kinetics of the channel [75][76].accumulation inside the mitochondria matrix, triggering excessive ROS production and the consequent higher sensitivity to apoptotic stress.
The characterization of MICU1
-/- mouse models have strengthened this concept and gave additional insight into the physiological role of this MCU regulator. In more detail, the characterization of MICU1-/- transgenic mice reveals that, despite partial postnatal mortality, the viable animals show marked ataxia and muscle weakness [77], a phenotype which is reminiscent of that of human patients bearing MICU1 genomic mutation [78]. Moreover, the MICU1−/− mice display several biochemical defects, including an increase in resting mitochondrial Catransgenic mice reveals that, despite partial postnatal mortality, the viable animals show marked ataxia and muscle weakness [41], a phenotype similar to that of human patients bearing MICU1 genomic mutation [42]. More recently, a new Ca
2+ levels, altered mitochondrial morphology, and a decreased ATP production [77]. Interestingly, the loss of MICU1 triggers a sustained pro-inflammatory response after partial hepatectomy and failure of liver regeneration in MICU1-deficient mice. In this scenario, the lack of MICU1 enhances mitochondrial permeability transition pore (PTP) opening in hepatocytes, thus leading to massive necrosis [79].-independent role of MICU1 in the regulation of cell metabolism via inhibition of the mitochondrial transport of pyruvate and fatty acids has emerged [43]. In addition, MICU1 is responsible for conferring and ensuring the stringent MCU selectivity for Ca
2+ and MCU core-complex composition [80]. This study reveals a mechanism that controls the MCU-mediated Caover Mn
2+ flux machinery and that relies on TCA cycle substrate availability. According to this view, the MICU1 regulatory axis acts as a metabolic homeostatic circuit to protect cells from the risk of bioenergetic crisis and mitochondrial Ca[44], which is crucial to guarantee the survival of cells sensitive to Mn
2+ overload during periods of nutrient stress. Altogether these findings [77][79][80] highlight once more the crucial importance of the fine-tuning of mitochondrial Casuch as neurons. Finally, super-resolution and electron microscopy studies showed that MICU1 also controls mitochondrial cristae junctions’ integrity [45].
Interestingly, a second variant of MICU1, named MICU1.1, has been discovered by our group as an alternative splicing product of the MICU1 mRNA [46] (Figure 1). MICU1.1 is well-conserved among species and expressed almost exclusively in skeletal muscle tissue and, to a less extent, in brain [46] (Figure 4). MICU1.1 behaves as a positive regulator of MCU, similarly and even more powerful than MICU1 due to its more efficient Ca
2+ uptake by Ca-binding, which leads to the activation of the uniporter at lower Ca
2+ and MICUs, especially in the context of promotion of cell survival under stress conditions.concentrations [46].
3.4. MICU1.12
3.5. MICU2
The mitochondrial calcium uptake protein 2 or MICU2, known also as EF-hand domain-containing family member A1 (EFHA1), was found to be a paralog of MICU1 [47] (Figure 1). MICU2 resides in the IMS, contains two EF-hand Ca
2+-binding domains, and interacts with both MICU1 and MCU [84] (Figure 2). The analysis of MICU2 transcript expression shown that this regulator has a peculiar cell-type distribution: it is present at high levels in the intestine, prostate, and cardiac tissues [84]. Biochemical data evidenced that MICU2 stability is strictly dependent on the presence of MICU1: indeed, silencing of MICU1 leads to loss of also MICU2 protein, while MICU1 (or MCU) overexpression stabilizes both MICUs in mammalian cells [84]. Moreover, the recently reported human MICU1-MICU2 crystal structure [45][85] revealed interesting details on the architecture of the heterodimer and of MICU2 in both the Ca-binding domains, and interacts with both MICU1 and MCU [47] (Figure 2). It is present at high levels in the intestine, prostate, and cardiac tissues and its stability strictly depends on the presence of MICU1 [47]. It has been shown that MICU2 positively regulates MCU activation by controlling the cytosolic [Ca
2+-bound and Ca] threshold for the relief of MICU1-mediated inhibition of MCU [48].
The MICU2 genetic ablation in mouse produces a decrease in the threshold for mitochondrial Ca
2+-free condition. The MICU1-MICU2 interaction sites have been identified and correspond to Glu242 in MICU1 and Arg352 in MICU2 in the Cauptake due to loss of the gatekeeping activity and overall loss of MCU-dependent Ca
2+-free state, while Phe383 in MICU1 and Glu196 in MICU2 contribute to the interaction of the two proteins in the Ca2+-bound state.influx due to the destabilization of the entire uniporter complex [49].
3.65. MICU3
MICU3, previously known as EFHA2, is another MICU1 paralog [47] (Figure 1) mainly expressed in the brain, much less expressed in skeletal muscle, and virtually absent in other tissues [50,51]. As the other MICUs, also MICU3 binds Ca
2+ thanks to the presence of EF-hand domains. The crystal structure of human MICU3 has been recently characterized in both Cathanks to the presence of EF-hand domains. The crystallographic analysis in both Ca
2+-free and Ca
2+-bound conditions [90]. This crystallographic analysis revealed a MICU3 3D structure very similar to that of MICU2, in line with the role of these factors as MCU channel gatekeepers at low intracellular Ca-bound conditions [52] revealed a MICU3 structure and function very similar to that of MICU2, in line with the role of these factors as MCU channel gatekeepers at low intracellular Ca
2+ levels. Upon cytosolic Calevels (Figures 3 and 4).
MICU3 functions as a potent positive regulator of mitochondrial Ca
2+ increase, MICU heterodimers, including those containing MICU3, undergo a conformational change that releases the latch formed upon the uniporter mouth, thus allowing Cauptake through heterodimerization with MICU1 [50]. The experimental evidence lead to hypothesize that the primary role of MICU3 is to enhance MCU channel activity in neurons to ensure efficient mitochondrial Ca
2+ flux through the MCU pore [90] (Figure 3 and Figure 4).uptake in response to both small and fast cytosolic Ca
2+ uptake through MICU1 [88]. Indeed, MICU3 forms heterodimers exclusively with MICU1, but not with MICU2, and the MICU1-MICU3 interaction leads to a significant increase of mitochondrial Ca2+ uptake, demonstrating the stimulatory action of MICU3 on uniporter activity [88]. Moreover, MICU3 downregulation blocks Ca2+ influx elicited by synaptic activity in primary cortical neurons, suggesting a specific role of this MCU regulator on neuronal function [88]. This line of evidence lets to hypothesize that the primary role of MICU3 is to enhance MCU opening to ensure mitochondrial Ca2+ uptake in response to both small and fast cytosolic Ca2+ rises, typical of synaptic neuronal stimulation (Figure 4).rises, typical of synaptic neuronal stimulation (Figure 4).
3.76. EMRE
The Essential MCU REgulator (EMRE) was discovered by
1). EMRE is a metazoan-specific protein of 10 kDa, ubiquitously expressed in all mammalian tissue, with one transmembrane domain, a mitochondrial targeting sequence, and a highly conserved C-terminus [91]. Moreover, it is required for the binding of MICU1 to MCU (Figure 2). Initial biochemical and cellular studies revealed that it is required for MCU function [91][92]. Indeed, in yeast cells, reconstituted with human MCU protein, the expression of MCU alone is not sufficient for uniporter activity, because the MCU channel is active only when also EMRE is co-expressed with the MCU pore-forming unit [92]. Interestingly, the knockdown of EMRE led to the loss of mitochondrial Caquantitative mass spectrometry analysis of affinity-purified MCU complex components [91] (Figure 1). EMRE is a metazoan-specific protein of 10 kDa, ubiquitously expressed in all mammalian tissue, with a single transmembrane domain [91]. It is required for the binding of MICU1 to MCU (Figure 2) and for MCU function [91,92]. EMRE was shown to control MCU activity by sensing Ca
2+ uptake to a similar extent to what was observed in MCU-silenced HEK-293T and HeLa cells [91]. The following studies tried to give a more detailed explanation of the EMRE function within the MCU complex [93]. EMRE was shown to control MCU activity by sensing Caelevation inside the matrix via its C-terminal domain and coordinating the other MCU regulators [93]. According to another model, EMRE N-terminus is present inside the matrix, while its C-terminal portion faces the IMS thus stimulating MCU activity via transmembrane helix interaction [94] and also by favoring MCU dimerization [53].
3.7. MCUR1
The CCDC90A gene, known as mitochondrial calcium uniporter regulator 1 or MCUR1, has been firstly identified from an RNAi screening searching for mitochondrial membrane components involved in mitochondrial Ca
uptake regulation [53] (Figure 1).
Only recently, crystallographic analysis of human MCUR1 protein revealed the sites of its interaction with MCU within the IMM. It has been shown that the MCUR1-dependent regulation of MCU depends on MCUR1 protein level and stability, similarly to what is reported for EMRE during MCU complex assembly [54].
Despite Paupe and collaborators claimed that MCUR1 is not a direct regulator of MCU, but rather a cytochrome c oxidase (COX) assembly factor [55] several lines of evidence suggest that it regulates the mitochondrial Ca
entry through MCU [53,56].
The latest findings assigned to MCUR1 also a relevant role in the control of cell metabolism [57] and autophagy [53,58,59] through modulation of mitochondrial Ca
. In mammalian cells, MCUR1 downregulation has been also linked to AMPK phosphorylation and LC3 processing [53], which directly links MCUR1 levels to the activation of autophagy [59].
The entry is from: 10.3390/biom11060786
4. References
- Raffaello, A.; Mammucari, C.; Gherardi, G.; Rizzuto, R. Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem. Sci. 2016, 41, 1035–1049, doi:10.1016/j.tibs.2016.09.001.
- Clapham, D.E. Calcium Signaling. Cell 2007, 131, 1047–1058, doi:10.1016/j.cell.2007.11.028.
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21.
- Rizzuto, R.; Pozzan, T. Microdomains of Intracellular Ca 2+ : Molecular Determinants and Functional Consequences. Physiol. Rev. 2006, 86, 369–408, doi:10.1152/physrev.00004.2005.
- Denton, R.M.; Randle, P.J.; Martin, B.R. Stimulation by calcium ions of pyruvate dehydrogenase phosphate phosphatase. Biochem. J. 1972, 128, 161–163, doi:10.1042/bj1280161.
- Denton, R.M.; Richards, D.A.; Chin, J.G. Calcium ions and the regulation of NAD+-linked isocitrate dehydrogenase from the mitochondria of rat heart and other tissues. Biochem. J. 1978, 176, 899–906, doi:10.1042/bj1760899.
- McCormack, J.G.; Denton, R.M. The effects of calcium ions and adenine nucleotides on the activity of pig heart 2-oxoglutarate dehydrogenase complex. Biochem. J. 1979, 180, 533–544, doi:10.1042/bj1800533.
- Territo, P.R.; Mootha, V.K.; French, S.A.; Balaban, R.S. Ca2+ activation of heart mitochondrial oxidative phosphorylation: Role of the F0/F1-ATPase. Am. J. Physiol. - Cell Physiol. 2000, 278, doi:10.1152/ajpcell.2000.278.2.c423.
- Glancy, B.; Willis, W.T.; Chess, D.J.; Balaban, R.S. Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry 2013, 52, 2793–2809, doi:10.1021/bi3015983.
- Ringer, S. A third contribution regarding the Influence of the Inorganic Constituents of the Blood on the Ventricular Contraction. J. Physiol. 1883, 4, 222–225, doi:10.1113/jphysiol.1883.sp000127.
- Slater, E.C.; Cleland, K.W. The effect of calcium on the respiratory and phosphorylative activities of heart-muscle sarcosomes. Biochem. J. 1953, 55, 566–580, doi:10.1042/bj0550566.
- Mitchell, P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 1961, 191, 144–148, doi:10.1038/191144a0.
- Stathopulos, P.B.; Ikura, M. Store operated calcium entry: From concept to structural mechanisms. Cell Calcium 2017, 63, 3–7.
- DeLuca, H.F.; Engstrom, G.W. Calcium uptake by rat kidney mitochondria. Proc. Natl. Acad. Sci. U. S. A. 1961, 47, 1744–1750, doi:10.1073/pnas.47.11.1744.
- Lehninger, A.L.; Rossi, C.S.; Greenawalt, J.W. Respiration-dependent accumulation of inorganic phosphate and Ca++ by rat liver mitochondria. Biochem. Biophys. Res. Commun. 1963, 10, 444–448, doi:10.1016/0006-291X(63)90377-2.
- Ridgway, E.B.; Ashley, C.C. Calcium transients in single muscle fibers. Biochem. Biophys. Res. Commun. 1967, 29, 229–234, doi:10.1016/0006-291X(67)90592-X.
- Rudolf, R.; Mongillo, M.; Rizzuto, R.; Pozzan, T. Looking forward to seeing calcium. Nat. Rev. Mol. Cell Biol. 2003, 4, 579–586, doi:10.1038/nrm1153.
- Takahashi, A.; Camacho, P.; Lechleiter, J.D.; Herman, B. Measurement of intracellular calcium. Physiol. Rev. 1999, 79, 1089–1125.
- Carafoli, E. The interplay of mitochondria with calcium: An historical appraisal. Cell Calcium 2012, 52, 1–8, doi:10.1016/j.ceca.2012.02.007.
- De Giorgi, F.; Brini, M.; Bastianutto, C.; Marsault, R.; Montero, M.; Pizzo, P.; Rossi, R.; Rizzuto, R. Targeting aequorin and green fluorescent protein to intracellular organelles. In Proceedings of the Gene; Elsevier B.V., 1996; Vol. 173, pp. 113–117.
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.; Fogarty, K.; Lifshitz, L.; Tuft, R.; Pozzan, T. Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca2+ Responses. Science (80-. ). 1998, 280, 1763–1766, doi:10.1126/science.280.5370.1763.
- Rizzuto, R.; Brini, M.; Murgia, M.; Pozzan, T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 1993, 262, 744–7.
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabò, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–40, doi:10.1038/nature10230.
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C. a; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345, doi:10.1038/nature10234.
- Kovács-Bogdán, E.; Sancak, Y.; Kamer, K.J.; Plovanich, M.; Jambhekar, A.; Huber, R.J.; Myre, M.A.; Blower, M.D.; Mootha, V.K. Reconstitution of the mitochondrial calcium uniporter in yeast. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 8985–90, doi:10.1073/pnas.1400514111.
- Perocchi, F.; Gohil, V.M.; Girgis, H.S.; Bao, X.R.; McCombs, J.E.; Palmer, A.E.; Mootha, V.K. MICU1 encodes a mitochondrial EF hand protein required for Ca2+uptake. Nature 2010, 467, 291–296, doi:10.1038/nature09358.
- Wang, Y.; Nguyen, N.X.; She, J.; Zeng, W.; Yang, Y.; Bai, X. chen; Jiang, Y. Structural Mechanism of EMRE-Dependent Gating of the Human Mitochondrial Calcium Uniporter. Cell 2019, 177, doi:10.1016/j.cell.2019.03.050.
- Zhuo, W.; Zhou, H.; Guo, R.; Yi, J.; Zhang, L.; Yu, L.; Sui, Y.; Zeng, W.; Wang, P.; Yang, M. Structure of intact human MCU supercomplex with the auxiliary MICU subunits. Protein Cell 2020.
- Wang, C.; Jacewicz, A.; Delgado, B.D.; Baradaran, R.; Long, S.B. Structures reveal gatekeeping of the mitochondrial Ca2+ uniporter by MICU1-MICU2. Elife 2020, 9, 1–30, doi:10.7554/eLife.59991.
- Fan, M.; Zhang, J.; Tsai, C.W.; Orlando, B.J.; Rodriguez, M.; Xu, Y.; Liao, M.; Tsai, M.F.; Feng, L. Structure and mechanism of the mitochondrial Ca2+ uniporter holocomplex. Nature 2020, doi:10.1038/s41586-020-2309-6.
- Pan, X.; Liu, J.; Nguyen, T.; Liu, C.; Sun, J.; Teng, Y.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. The Physiological Role of Mitochondrial Calcium Revealed by Mice Lacking the Mitochondrial Calcium Uniporter. Nat. Cell Biol. 2013, 15, 1464–1472, doi:10.1038/ncb2868.
- Huang, G.; Vercesi, A.E.; Docampo, R. Essential regulation of cell bioenergetics in Trypanosoma brucei by the mitochondrial calcium uniporter. Nat. Commun. 2013, doi:10.1038/ncomms3865.
- Prudent, J.; Popgeorgiev, N.; Bonneau, B.; Thibaut, J.; Gadet, R.; Lopez, J.; Gonzalo, P.; Rimokh, R.; Manon, S.; Houart, C.; et al. Bcl-wav and the mitochondrial calcium uniporter drive gastrula morphogenesis in zebrafish. Nat. Commun. 2013, doi:10.1038/ncomms3330.
- Murphy, E.; Pan, X.; Nguyen, T.; Liu, J.; Holmström, K.M.; Finkel, T. Unresolved questions from the analysis of mice lacking MCU expression. Biochem. Biophys. Res. Commun. 2014, 449, 384–385, doi:10.1016/j.bbrc.2014.04.144.
- Drago, I.; Davis, R.L. Inhibiting the Mitochondrial Calcium Uniporter during Development Impairs Memory in Adult Drosophila. Cell Rep. 2016, doi:10.1016/j.celrep.2016.08.017.
- Raffaello, A.; De Stefani, D.; Sabbadin, D.; Teardo, E.; Merli, G.; Picard, A.; Checchetto, V.; Moro, S.; Szabò, I.; Rizzuto, R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013, 32157, 2362–2376, doi:10.1038/emboj.2013.157.
- Chiurillo, M.A.; Lander, N.; Bertolini, M.S.; Storey, M.; Vercesi, A.E.; Docampo, R. Different Roles of Mitochondrial Calcium Uniporter Complex Subunits in Growth and Infectivity of Trypanosoma cruzi. MBio 2017, 8, e00574-17, doi:10.1128/mBio.00574-17.
- Fieni, F.; Lee, S.B.; Jan, Y.N.; Kirichok, Y. Activity of the mitochondrial calcium uniporter varies greatly between tissues. Nat. Commun. 2012, 3, 1317, doi:10.1038/ncomms2325.
- Mammucari, C.; Raffaello, A.; Vecellio Reane, D.; Gherardi, G.; De Mario, A.; Rizzuto, R. Mitochondrial calcium uptake in organ physiology: from molecular mechanism to animal models. Pflügers Arch. - Eur. J. Physiol. 2018, 470, 1165–1179, doi:10.1007/s00424-018-2123-2.
- Csordás, G.; Golenár, T.; Seifert, E.L.; Kamer, K.J.; Sancak, Y.; Perocchi, F.; Moffat, C.; Weaver, D.; Perez, S.D.L.F.; Bogorad, R.; et al. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca2+ uniporter. Cell Metab. 2013, 17, 976–987, doi:10.1016/j.cmet.2013.04.020.
- Liu, J.C.; Liu, J.; Holmström, K.M.; Menazza, S.; Parks, R.J.; Fergusson, M.M.; Yu, Z.X.; Springer, D.A.; Halsey, C.; Liu, C.; et al. MICU1 Serves as a Molecular Gatekeeper to Prevent In Vivo Mitochondrial Calcium Overload. Cell Rep. 2016, 16, 1561–1573, doi:10.1016/j.celrep.2016.07.011.
- Logan, C. V; Szabadkai, G.; Sharpe, J.A.; Parry, D.A.; Torelli, S.; Childs, A.-M.; Kriek, M.; Phadke, R.; Johnson, C.A.; Roberts, N.Y.; et al. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat. Genet. 2014, 46, 188–93, doi:10.1038/ng.2851.
- Nemani, N.; Dong, Z.; Daw, C.C.; Madaris, T.R.; Ramachandran, K.; Enslow, B.T.; Rubannelsonkumar, C.S.; Shanmughapriya, S.; Mallireddigari, V.; Maity, S.; et al. Mitochondrial pyruvate and fatty acid flux modulate MICU1-dependent control of MCU activity. Sci. Signal. 2020, 13, 6206, doi:10.1126/scisignal.aaz6206.
- Kamer, K.J.; Sancak, Y.; Fomina, Y.; Meisel, J.D.; Chaudhuri, D.; Grabarek, Z.; Mootha, V.K. MICU1 imparts the mitochondrial uniporter with the ability to discriminate between Ca2+ and Mn2+. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E7960–E7969, doi:10.1073/pnas.1807811115.
- Gottschalk, B.; Klec, C.; Leitinger, G.; Bernhart, E.; Rost, R.; Bischof, H.; Madreiter-Sokolowski, C.T.; Radulović, S.; Eroglu, E.; Sattler, W.; et al. MICU1 controls cristae junction and spatially anchors mitochondrial Ca2+ uniporter complex. Nat. Commun. 2019, doi:10.1038/s41467-019-11692-x.
- Vecellio Reane, D.; Vallese, F.; Checchetto, V.; Acquasaliente, L.; Butera, G.; De Filippis, V.; Szabò, I.; Zanotti, G.; Rizzuto, R.; Raffaello, A. A MICU1 Splice Variant Confers High Sensitivity to the Mitochondrial Ca2+ Uptake Machinery of Skeletal Muscle. Mol. Cell 2016, 64, 760–773, doi:10.1016/j.molcel.2016.10.001.
- Plovanich, M.; Bogorad, R.L.; Sancak, Y.; Kamer, K.J.; Strittmatter, L.; Li, A.A.; Girgis, H.S.; Kuchimanchi, S.; De Groot, J.; Speciner, L.; et al. MICU2, a Paralog of MICU1, Resides within the Mitochondrial Uniporter Complex to Regulate Calcium Handling. PLoS One 2013, 8, doi:10.1371/journal.pone.0055785.
- Payne, R.; Hoff, H.; Roskowski, A.; Foskett Correspondence, J.K.; Foskett, J.K. MICU2 Restricts Spatial Crosstalk between InsP 3 R and MCU Channels by Regulating Threshold and Gain of MICU1-Mediated Inhibition and Activation of MCU. Cell Rep. 2017, 21, 3141–3154, doi:10.1016/j.celrep.2017.11.064.
- Bick, A.G.; Wakimoto, H.; Kamer, K.J.; Sancak, Y.; Goldberger, O.; Axelsson, A.; DeLaughter, D.M.; Gorham, J.M.; Mootha, V.K.; Seidman, J.G.; et al. Cardiovascular homeostasis dependence on MICU2, a regulatory subunit of the mitochondrial calcium uniporter. Proc. Natl. Acad. Sci. 2017, 114, 201711303, doi:10.1073/pnas.1711303114.
- Patron, M.; Granatiero, V.; Espino, J.; Rizzuto, R.; De Stefani, D. MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ. 2018, doi:10.1038/s41418-018-0113-8.
- Márkus, N.M.; Hasel, P.; Qiu, J.; Bell, K.F.S.; Heron, S.; Kind, P.C.; Dando, O.; Simpson, T.I.; Hardingham, G.E. Expression of mRNA encoding Mcu and other mitochondrial calcium regulatory genes depends on cell type, neuronal subtype, and ca2+ signaling. PLoS One 2016, doi:10.1371/journal.pone.0148164.
- Xing, Y.; Wang, M.; Wang, J.; Nie, Z.; Wu, G.; Yang, X.; Shen, Y. Dimerization of MICU Proteins Controls Ca2+ Influx through the Mitochondrial Ca2+ Uniporter. Cell Rep. 2019, doi:10.1016/j.celrep.2019.01.022.
- Mallilankaraman, K.; Cárdenas, C.; Doonan, P.J.; Chandramoorthy, H.C.; Irrinki, K.M.; Golenár, T.; Csordás, G.; Madireddi, P.; Yang, J.; Müller, M.; et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat. Cell Biol. 2012, 14, 1336–1343, doi:10.1038/ncb2622.
- Adlakha, J.; Karamichali, I.; Sangwallek, J.; Deiss, S.; Bär, K.; Coles, M.; Hartmann, M.D.; Lupas, A.N.; Hernandez Alvarez, B. Characterization of MCU-Binding Proteins MCUR1 and CCDC90B — Representatives of a Protein Family Conserved in Prokaryotes and Eukaryotic Organelles. Structure 2019, doi:10.1016/j.str.2018.11.004.
- Paupe, V.; Prudent, J.; Dassa, E.P.; Rendon, O.Z.; Shoubridge, E.A. CCDC90A (MCUR1) is a cytochrome c oxidase assembly factor and not a regulator of the mitochondrial calcium uniporter. Cell Metab. 2015, 21, 109–116, doi:10.1016/j.cmet.2014.12.004.
- Vais, H.; Tanis, J.E.; Müller, M.; Payne, R.; Mallilankaraman, K.; Foskett, J.K. MCUR1, CCDC90A, is a regulator of the mitochondrial calcium uniporter. Cell Metab. 2015, 22, 533–535.
- Zulkifli, M.; Neff, J.K.; Timbalia, S.A.; Garza, N.M.; Chen, Y.; Watrous, J.D.; Murgia, M.; Trivedi, P.P.; Anderson, S.K.; Tomar, D.; et al. Yeast homologs of human MCUR1 regulate mitochondrial proline metabolism. Nat. Commun. 2020, doi:10.1038/s41467-020-18704-1.
- Chaudhuri, D.; Artiga, D.J.; Abiria, S.A.; Clapham, D.E. Mitochondrial calcium uniporter regulator 1 (MCUR1) regulates the calcium threshold for the mitochondrial permeability transition. Proc. Natl. Acad. Sci. 2016, 113, E1872–E1880, doi:10.1073/pnas.1602264113.
- Natarajan, V.; Mah, T.; Peishi, C.; Tan, S.Y.; Chawla, R.; Arumugam, T.V.; Ramasamy, A.; Mallilankaraman, K. Oxygen Glucose Deprivation Induced Prosurvival Autophagy Is Insufficient to Rescue Endothelial Function. Front. Physiol. 2020, doi:10.3389/fphys.2020.533683.