 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Stephanie Hehlgans | + 2777 word(s) | 2777 | 2021-06-07 06:08:47 | | | |
| 2 | Nora Tang | Meta information modification | 2777 | 2021-06-25 08:59:11 | | |
Video Upload Options
Tumor suppressor 53 (p53) is a multifunctional protein that regulates cell cycle, DNA repair, apoptosis and metabolic pathways. In colorectal cancer (CRC), mutations of the gene occur in 60% of patients and are associated with a more aggressive tumor phenotype and resistance to anti-cancer therapy. In addition, inhibitor of apoptosis (IAP) proteins are distinguished biomarkers overexpressed in CRC that impact on a diverse set of signaling pathways associated with the regulation of apoptosis/autophagy, cell migration, cell cycle and DNA damage response. As these mechanisms are further firmly controlled by p53, a transcriptional and post-translational regulation of IAPs by p53 is expected to occur in cancer cells. Here, we aim to review the molecular regulatory mechanisms between IAPs and p53 and discuss the therapeutic potential of targeting their interrelationship by multimodal treatment options.
1. Introduction
2. Biology and Functions of p53, a Brief Introduction
3. Structure and Function of the Inhibitor of Apoptosis Protein Family (IAP)
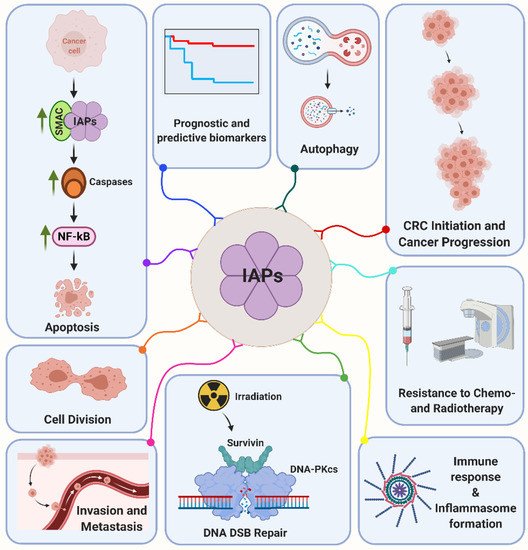
3.1. cIAP1 and cIAP2
3.2. XIAP
3.3. Survivin
3.4. BRUCE/Apollon
3.5. LIVIN
3.6. NAIP and hILP-2
References
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480.
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Nakamura, Y.; White, R.; Smits, A.M.; Bos, J.L. Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 1988, 319, 525–532.
- Simon, K. Colorectal cancer development and advances in screening. Clin. Interv. Aging 2016, 11, 967–976.
- Nakayama, M.; Oshima, M. Mutant p53 in colon cancer. J. Mol. Cell Biol. 2019, 11, 267–276.
- Solomon, H.; Dinowitz, N.; Pateras, I.S.; Cooks, T.; Shetzer, Y.; Molchadsky, A.; Charni, M.; Rabani, S.; Koifman, G.; Tarcic, O.; et al. Mutant p53 gain of function underlies high expression levels of colorectal cancer stem cells markers. Oncogene 2018, 37, 1669–1684.
- Schulz-Heddergott, R.; Moll, U.M. Gain-of-Function (GOF) Mutant p53 as Actionable Therapeutic Target. Cancers 2018, 10, 188.
- Van Gijn, W.; Marijnen, C.A.; Nagtegaal, I.D.; Kranenbarg, E.M.; Putter, H.; Wiggers, T.; Rutten, H.J.; Pahlman, L.; Glimelius, B.; van de Velde, C.J. Preoperative radiotherapy combined with total mesorectal excision for resectable rectal cancer: 12-year follow-up of the multicentre, randomised controlled TME trial. Lancet Oncol. 2011, 12, 575–582.
- Rodel, C.; Liersch, T.; Becker, H.; Fietkau, R.; Hohenberger, W.; Hothorn, T.; Graeven, U.; Arnold, D.; Lang-Welzenbach, M.; Raab, H.R.; et al. Preoperative chemoradiotherapy and postoperative chemotherapy with fluorouracil and oxaliplatin versus fluorouracil alone in locally advanced rectal cancer: Initial results of the German CAO/ARO/AIO-04 randomised phase 3 trial. Lancet Oncol. 2012, 13, 679–687.
- Fokas, E.; Glynne-Jones, R.; Appelt, A.; Beets-Tan, R.; Beets, G.; Haustermans, K.; Marijnen, C.; Minsky, B.D.; Ludmir, E.; Quirke, P.; et al. Outcome measures in multimodal rectal cancer trials. Lancet Oncol. 2020, 21, e252–e264.
- Fokas, E.; Fietkau, R.; Hartmann, A.; Hohenberger, W.; Grutzmann, R.; Ghadimi, M.; Liersch, T.; Strobel, P.; Grabenbauer, G.G.; Graeven, U.; et al. Neoadjuvant rectal score as individual-level surrogate for disease-free survival in rectal cancer in the CAO/ARO/AIO-04 randomized phase III trial. Ann. Oncol. 2018, 29, 1521–1527.
- Fokas, E.; Strobel, P.; Fietkau, R.; Ghadimi, M.; Liersch, T.; Grabenbauer, G.G.; Hartmann, A.; Kaufmann, M.; Sauer, R.; Graeven, U.; et al. Tumor Regression Grading After Preoperative Chemoradiotherapy as a Prognostic Factor and Individual-Level Surrogate for Disease-Free Survival in Rectal Cancer. J. Natl. Cancer Inst. 2017, 109, djx095.
- Mohamed, M.S.; Bishr, M.K.; Almutairi, F.M.; Ali, A.G. Inhibitors of apoptosis: Clinical implications in cancer. Apoptosis 2017, 22, 1487–1509.
- Alvarez-Gonzalez, R. Genomic maintenance: The p53 poly(ADP-ribosyl)ation connection. Sci. Signal. 2007, 2007, pe68.
- Gajewski, S.; Hartwig, A. PARP1 Is Required for ATM-Mediated p53 Activation and p53-Mediated Gene Expression after Ionizing Radiation. Chem. Res. Toxicol. 2020, 33, 1933–1940.
- Christmann, M.; Kaina, B. Epigenetic regulation of DNA repair genes and implications for tumor therapy. Mutat. Res. 2019, 780, 15–28.
- Joerger, A.C.; Fersht, A.R. The tumor suppressor p53: From structures to drug discovery. Cold Spring Harb. Perspect. Biol. 2010, 2, a000919.
- Brazda, V.; Fojta, M. The Rich World of p53 DNA Binding Targets: The Role of DNA Structure. Int. J. Mol. Sci. 2019, 20, 5605.
- Inoue, K.; Fry, E.A.; Frazier, D.P. Transcription factors that interact with p53 and Mdm2. Int. J. Cancer 2016, 138, 1577–1585.
- Murray, D.; Mirzayans, R. Cellular Responses to Platinum-Based Anticancer Drugs and UVC: Role of p53 and Implications for Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 5766.
- Dobbelstein, M.; Levine, A.J. Mdm2: Open questions. Cancer Sci. 2020, 111, 2203–2211.
- Achanta, G.; Pelicano, H.; Feng, L.; Plunkett, W.; Huang, P. Interaction of p53 and DNA-PK in response to nucleoside analogues: Potential role as a sensor complex for DNA damage. Cancer Res. 2001, 61, 8723–8729.
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412.
- Birnbaum, M.J.; Clem, R.J.; Miller, L.K. An apoptosis-inhibiting gene from a nuclear polyhedrosis virus encoding a polypeptide with Cys/His sequence motifs. J. Virol. 1994, 68, 2521–2528.
- Crook, N.E.; Clem, R.J.; Miller, L.K. An apoptosis-inhibiting baculovirus gene with a zinc finger-like motif. J. Virol. 1993, 67, 2168–2174.
- Miura, K.; Fujibuchi, W.; Ishida, K.; Naitoh, T.; Ogawa, H.; Ando, T.; Yazaki, N.; Watanabe, K.; Haneda, S.; Shibata, C.; et al. Inhibitor of apoptosis protein family as diagnostic markers and therapeutic targets of colorectal cancer. Surg. Today 2011, 41, 175–182.
- Hrdinka, M.; Yabal, M. Inhibitor of apoptosis proteins in human health and disease. Genes Immun. 2019, 20, 641–650.
- Ng, A.; Xavier, R.J. Leucine-rich repeat (LRR) proteins: Integrators of pattern recognition and signaling in immunity. Autophagy 2011, 7, 1082–1084.
- LaCasse, E.C.; Baird, S.; Korneluk, R.G.; MacKenzie, A.E. The inhibitors of apoptosis (IAPs) and their emerging role in cancer. Oncogene 1998, 17, 3247–3259.
- Rothe, M.; Pan, M.G.; Henzel, W.J.; Ayres, T.M.; Goeddel, D.V. The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell 1995, 83, 1243–1252.
- Busca, A.; Konarski, Y.; Gajanayaka, N.; O’Hara, S.; Angel, J.; Kozlowski, M.; Kumar, A. cIAP1/2-TRAF2-SHP-1-Src-MyD88 Complex Regulates Lipopolysaccharide-Induced IL-27 Production through NF-kappaB Activation in Human Macrophages. J. Immunol. 2018, 200, 1593–1606.
- Mao, A.P.; Li, S.; Zhong, B.; Li, Y.; Yan, J.; Li, Q.; Teng, C.; Shu, H.B. Virus-triggered ubiquitination of TRAF3/6 by cIAP1/2 is essential for induction of interferon-beta (IFN-beta) and cellular antiviral response. J. Biol. Chem. 2010, 285, 9470–9476.
- Huang, L.-G.; Li, J.-P.; Pang, X.-M.; Chen, C.-Y.; Xiang, H.-Y.; Feng, L.-B.; Su, S.-Y.; Li, S.-H.; Zhang, L.; Liu, J.-L. MicroRNA-29c Correlates with Neuroprotection Induced by FNS by Targeting Both Birc2 and Bak1 in Rat Brain after Stroke. CNS Neurosci. Ther. 2015, 21, 496–503.
- Goncharov, T.; Niessen, K.; de Almagro, M.C.; Izrael-Tomasevic, A.; Fedorova, A.V.; Varfolomeev, E.; Arnott, D.; Deshayes, K.; Kirkpatrick, D.S.; Vucic, D. OTUB1 modulates c-IAP1 stability to regulate signalling pathways. EMBO J. 2013, 32, 1103–1114.
- Tenev, T.; Bianchi, K.; Darding, M.; Broemer, M.; Langlais, C.; Wallberg, F.; Zachariou, A.; Lopez, J.; MacFarlane, M.; Cain, K.; et al. The Ripoptosome, a Signaling Platform that Assembles in Response to Genotoxic Stress and Loss of IAPs. Mol. Cell 2011, 43, 432–448.
- Nicholson, J.; Jevons, S.J.; Groselj, B.; Ellermann, S.; Konietzny, R.; Kerr, M.; Kessler, B.M.; Kiltie, A.E. E3 Ligase cIAP2 Mediates Downregulation of MRE11 and Radiosensitization in Response to HDAC Inhibition in Bladder Cancer. Cancer Res. 2017, 77, 3027–3039.
- Nudel, K.; Massari, P.; Genco, C.A. Neisseria gonorrhoeae Modulates Cell Death in Human Endocervical Epithelial Cells through Export of Exosome-Associated cIAP2. Infect. Immun. 2015, 83, 3410–3417.
- Valenzuela, M.M.; Ferguson Bennit, H.R.; Gonda, A.; Diaz Osterman, C.J.; Hibma, A.; Khan, S.; Wall, N.R. Exosomes Secreted from Human Cancer Cell Lines Contain Inhibitors of Apoptosis (IAP). Cancer Microenviron. 2015, 8, 65–73.
- Khan, S.; Bennit, H.F.; Wall, N.R. The emerging role of exosomes in survivin secretion. Histol. Histopathol. 2015, 30, 43–50.
- Duckett, C.S.; Nava, V.E.; Gedrich, R.W.; Clem, R.J.; Van Dongen, J.L.; Gilfillan, M.C.; Shiels, H.; Hardwick, J.M.; Thompson, C.B. A conserved family of cellular genes related to the baculovirus iap gene and encoding apoptosis inhibitors. EMBO J. 1996, 15, 2685–2694.
- Silke, J.; Vucic, D. IAP family of cell death and signaling regulators. Methods Enzymol. 2014, 545, 35–65.
- Xu, J.; Hua, X.; Yang, R.; Jin, H.; Li, J.; Zhu, J.; Tian, Z.; Huang, M.; Jiang, G.; Huang, H.; et al. XIAP Interaction with E2F1 and Sp1 via its BIR2 and BIR3 domains specific activated MMP2 to promote bladder cancer invasion. Oncogenesis 2019, 8, 71.
- Delbue, D.; Mendonca, B.S.; Robaina, M.C.; Lemos, L.G.T.; Lucena, P.I.; Viola, J.P.B.; Magalhaes, L.M.; Crocamo, S.; Oliveira, C.A.B.; Teixeira, F.R.; et al. Expression of nuclear XIAP associates with cell growth and drug resistance and confers poor prognosis in breast cancer. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118761.
- Wicki, S.; Gurzeler, U.; Wei-Lynn Wong, W.; Jost, P.J.; Bachmann, D.; Kaufmann, T. Loss of XIAP facilitates switch to TNFalpha-induced necroptosis in mouse neutrophils. Cell Death Dis. 2016, 7, e2422.
- Liao, Y.; Zhao, J.; Bulek, K.; Tang, F.; Chen, X.; Cai, G.; Jia, S.; Fox, P.L.; Huang, E.; Pizarro, T.T.; et al. Inflammation mobilizes copper metabolism to promote colon tumorigenesis via an IL-17-STEAP4-XIAP axis. Nat. Commun. 2020, 11, 900.
- Zilu, S.; Qian, H.; Haibin, W.; Chenxu, G.; Deshuai, L.; Qiang, L.; Linfeng, H.; Jun, T.; Minxuan, X. Effects of XIAP on high fat diet-induced hepatic steatosis: A mechanism involving NLRP3 inflammasome and oxidative stress. Aging 2019, 11, 12177–12201.
- Lu, M.; Qin, X.; Yao, J.; Yang, Y.; Zhao, M.; Sun, L. MiR-134-5p targeting XIAP modulates oxidative stress and apoptosis in cardiomyocytes under hypoxia/reperfusion-induced injury. IUBMB Life 2020, 72, 2154–2166.
- Damgaard, R.B.; Nachbur, U.; Yabal, M.; Wong, W.W.; Fiil, B.K.; Kastirr, M.; Rieser, E.; Rickard, J.A.; Bankovacki, A.; Peschel, C.; et al. The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol. Cell 2012, 46, 746–758.
- Andree, M.; Seeger, J.M.; Schull, S.; Coutelle, O.; Wagner-Stippich, D.; Wiegmann, K.; Wunderlich, C.M.; Brinkmann, K.; Broxtermann, P.; Witt, A.; et al. BID-dependent release of mitochondrial SMAC dampens XIAP-mediated immunity against Shigella. EMBO J. 2014, 33, 2171–2187.
- Huang, X.; Wu, Z.; Mei, Y.; Wu, M. XIAP inhibits autophagy via XIAP-Mdm2-p53 signalling. EMBO J. 2013, 32, 2204–2216.
- Lin, F.; Ghislat, G.; Luo, S.; Renna, M.; Siddiqi, F.; Rubinsztein, D.C. XIAP and cIAP1 amplifications induce Beclin 1-dependent autophagy through NFkappaB activation. Hum. Mol. Genet. 2015, 24, 2899–2913.
- Gyrd-Hansen, M.; Meier, P. IAPs: From caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat. Rev. Cancer 2010, 10, 561–574.
- Jin, H.S.; Lee, D.H.; Kim, D.H.; Chung, J.H.; Lee, S.J.; Lee, T.H. cIAP1, cIAP2, and XIAP act cooperatively via nonredundant pathways to regulate genotoxic stress-induced nuclear factor-kappaB activation. Cancer Res. 2009, 69, 1782–1791.
- Moussata, D.; Amara, S.; Siddeek, B.; Decaussin, M.; Hehlgans, S.; Paul-Bellon, R.; Mornex, F.; Gerard, J.P.; Romestaing, P.; Rodel, F.; et al. XIAP as a radioresistance factor and prognostic marker for radiotherapy in human rectal adenocarcinoma. Am. J. Pathol. 2012, 181, 1271–1278.
- Hehlgans, S.; Petraki, C.; Reichert, S.; Cordes, N.; Rodel, C.; Rodel, F. Double targeting of Survivin and XIAP radiosensitizes 3D grown human colorectal tumor cells and decreases migration. Radiother. Oncol. 2013, 108, 32–39.
- Ambrosini, G.; Adida, C.; Altieri, D.C. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat. Med. 1997, 3, 917–921.
- Altieri, D.C. Validating survivin as a cancer therapeutic target. Nat. Rev. Cancer 2003, 3, 46–54.
- Barrera-Vazquez, O.S.; Cancio-Lonches, C.; Hernandez-Gonzalez, O.; Chavez-Munguia, B.; Villegas-Sepulveda, N.; Gutierrez-Escolano, A.L. The feline calicivirus leader of the capsid protein causes survivin and XIAP downregulation and apoptosis. Virology 2019, 527, 146–158.
- Eckelman, B.P.; Salvesen, G.S.; Scott, F.L. Human inhibitor of apoptosis proteins: Why XIAP is the black sheep of the family. EMBO Rep. 2006, 7, 988–994.
- Li, F.; Ambrosini, G.; Chu, E.Y.; Plescia, J.; Tognin, S.; Marchisio, P.C.; Altieri, D.C. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature 1998, 396, 580–584.
- Vader, G.; Kauw, J.J.W.; Medema, R.H.; Lens, S.M.A. Survivin mediates targeting of the chromosomal passenger complex to the centromere and midbody. EMBO Rep. 2006, 7, 85–92.
- Wang, H.; Holloway, M.P.; Ma, L.; Cooper, Z.A.; Riolo, M.; Samkari, A.; Elenitoba-Johnson, K.S.; Chin, Y.E.; Altura, R.A. Acetylation directs survivin nuclear localization to repress STAT3 oncogenic activity. J. Biol. Chem. 2010, 285, 36129–36137.
- Engelsma, D.; Rodriguez, J.A.; Fish, A.; Giaccone, G.; Fornerod, M. Homodimerization antagonizes nuclear export of survivin. Traffic 2007, 8, 1495–1502.
- Wheatley, S.P.; Altieri, D.C. Survivin at a glance. J. Cell Sci. 2019, 132.
- Lu, B.; Mu, Y.; Cao, C.; Zeng, F.; Schneider, S.; Tan, J.; Price, J.; Chen, J.; Freeman, M.; Hallahan, D.E. Survivin as a therapeutic target for radiation sensitization in lung cancer. Cancer Res. 2004, 64, 2840–2845.
- Chakravarti, A.; Zhai, G.G.; Zhang, M.; Malhotra, R.; Latham, D.E.; Delaney, M.A.; Robe, P.; Nestler, U.; Song, Q.; Loeffler, J. Survivin enhances radiation resistance in primary human glioblastoma cells via caspase-independent mechanisms. Oncogene 2004, 23, 7494–7506.
- Rodel, F.; Hoffmann, J.; Distel, L.; Herrmann, M.; Noisternig, T.; Papadopoulos, T.; Sauer, R.; Rodel, C. Survivin as a radioresistance factor, and prognostic and therapeutic target for radiotherapy in rectal cancer. Cancer Res. 2005, 65, 4881–4887.
- Capalbo, G.; Dittmann, K.; Weiss, C.; Reichert, S.; Hausmann, E.; Rodel, C.; Rodel, F. Radiation-induced survivin nuclear accumulation is linked to DNA damage repair. Int. J. Radiat. Oncol. Biol. Phys. 2010, 77, 226–234.
- Wang, X.; Beitler, J.J.; Huang, W.; Chen, G.; Qian, G.; Magliocca, K.; Patel, M.R.; Chen, A.Y.; Zhang, J.; Nannapaneni, S.; et al. Honokiol Radiosensitizes Squamous Cell Carcinoma of the Head and Neck by Downregulation of Survivin. Clin. Cancer Res. 2018, 24, 858–869.
- Gullulu, O.; Hehlgans, S.; Mayer, B.E.; Gossner, I.; Petraki, C.; Hoffmann, M.; Dombrowsky, M.J.; Kunzmann, P.; Hamacher, K.; Strebhardt, K.; et al. A spatial and functional interaction of a heterotetramer Survivin-DNA-PKcs complex in DNA damage response. Cancer Res. 2021.
- Hauser, H.P.; Bardroff, M.; Pyrowolakis, G.; Jentsch, S. A giant ubiquitin-conjugating enzyme related to IAP apoptosis inhibitors. J. Cell Biol. 1998, 141, 1415–1422.
- Hitz, C.; Vogt-Weisenhorn, D.; Ruiz, P.; Wurst, W.; Floss, T. Progressive loss of the spongiotrophoblast layer of Birc6/Bruce mutants results in embryonic lethality. Genesis 2005, 42, 91–103.
- Hao, Y.; Sekine, K.; Kawabata, A.; Nakamura, H.; Ishioka, T.; Ohata, H.; Katayama, R.; Hashimoto, C.; Zhang, X.; Noda, T.; et al. Apollon ubiquitinates SMAC and caspase-9, and has an essential cytoprotection function. Nat. Cell Biol. 2004, 6, 849–860.
- Sekine, K.; Hao, Y.; Suzuki, Y.; Takahashi, R.; Tsuruo, T.; Naito, M. HtrA2 cleaves Apollon and induces cell death by IAP-binding motif in Apollon-deficient cells. Biochem. Biophys. Res. Commun. 2005, 330, 279–285.
- Kikuchi, R.; Ohata, H.; Ohoka, N.; Kawabata, A.; Naito, M. APOLLON protein promotes early mitotic CYCLIN A degradation independent of the spindle assembly checkpoint. J. Biol. Chem. 2014, 289, 3457–3467.
- Pohl, C.; Jentsch, S. Final stages of cytokinesis and midbody ring formation are controlled by BRUCE. Cell 2008, 132, 832–845.
- Ge, C.; Che, L.; Ren, J.; Pandita, R.K.; Lu, J.; Li, K.; Pandita, T.K.; Du, C. BRUCE regulates DNA double-strand break response by promoting USP8 deubiquitination of BRIT1. Proc. Natl. Acad. Sci. USA 2015, 112, E1210–E1219.
- Ge, C.; Vilfranc, C.L.; Che, L.; Pandita, R.K.; Hambarde, S.; Andreassen, P.R.; Niu, L.; Olowokure, O.; Shah, S.; Waltz, S.E.; et al. The BRUCE-ATR Signaling Axis Is Required for Accurate DNA Replication and Suppression of Liver Cancer Development. Hepatology 2019, 69, 2608–2622.
- Sun, J.G.; Liao, R.X.; Zhang, S.X.; Duan, Y.Z.; Zhuo, W.L.; Wang, X.X.; Wang, Z.X.; Li, D.Z.; Chen, Z.T. Role of inhibitor of apoptosis protein Livin in radiation resistance in nonsmall cell lung cancer. Cancer Biother. Radiopharm. 2011, 26, 585–592.
- Wu, S.Q.; Xu, Q.B.; Sheng, W.Y.; Su, L.Y.; Zhu, L.W. A novel role for Livin in the response to ultraviolet B radiation and pterygium development. Int. J. Mol. Med. 2020, 45, 1103–1111.
- Chen, F.; Yang, D.; Wang, S.; Che, X.; Wang, J.; Li, X.; Zhang, Z.; Chen, X.; Song, X. Livin regulates prostate cancer cell invasion by impacting the NF-kappaB signaling pathway and the expression of FN and CXCR4. IUBMB Life 2012, 64, 274–283.
- Li, F.; Yin, X.; Luo, X.; Li, H.Y.; Su, X.; Wang, X.Y.; Chen, L.; Zheng, K.; Ren, G.S. Livin promotes progression of breast cancer through induction of epithelial-mesenchymal transition and activation of AKT signaling. Cell Signal. 2013, 25, 1413–1422.
- Hsieh, C.H.; Lin, Y.J.; Wu, C.P.; Lee, H.T.; Shyu, W.C.; Wang, C.C. Livin contributes to tumor hypoxia-induced resistance to cytotoxic therapies in glioblastoma multiforme. Clin. Cancer Res. 2015, 21, 460–470.
- Kasof, G.M.; Gomes, B.C. Livin, a novel inhibitor of apoptosis protein family member. J. Biol. Chem. 2001, 276, 3238–3246.
- Nachmias, B.; Ashhab, Y.; Bucholtz, V.; Drize, O.; Kadouri, L.; Lotem, M.; Peretz, T.; Mandelboim, O.; Ben-Yehuda, D. Caspase-mediated cleavage converts Livin from an antiapoptotic to a proapoptotic factor: Implications for drug-resistant melanoma. Cancer Res. 2003, 63, 6340–6349.
- Nachmias, B.; Lazar, I.; Elmalech, M.; Abed-El-Rahaman, I.; Asshab, Y.; Mandelboim, O.; Perlman, R.; Ben-Yehuda, D. Subcellular localization determines the delicate balance between the anti- and pro-apoptotic activity of Livin. Apoptosis 2007, 12, 1129–1142.
- Wang, X.; Xu, J.; Ju, S.; Ni, H.; Zhu, J.; Wang, H. Livin gene plays a role in drug resistance of colon cancer cells. Clin. Biochem. 2010, 43, 655–660.
- Oh, B.Y.; Lee, R.A.; Kim, K.H. siRNA targeting Livin decreases tumor in a xenograft model for colon cancer. World J. Gastroenterol. 2011, 17, 2563–2571.
- Roy, N.; Mahadevan, M.S.; McLean, M.; Shutler, G.; Yaraghi, Z.; Farahani, R.; Baird, S.; Besner-Johnston, A.; Lefebvre, C.; Kang, X.; et al. The gene for neuronal apoptosis inhibitory protein is partially deleted in individuals with spinal muscular atrophy. Cell 1995, 80, 167–178.
- Abadia-Molina, F.; Moron-Calvente, V.; Baird, S.D.; Shamim, F.; Martin, F.; MacKenzie, A. Neuronal apoptosis inhibitory protein (NAIP) localizes to the cytokinetic machinery during cell division. Sci. Rep. 2017, 7, 39981.
- Karki, R.; Lee, E.; Place, D.; Samir, P.; Mavuluri, J.; Sharma, B.R.; Balakrishnan, A.; Malireddi, R.K.S.; Geiger, R.; Zhu, Q.; et al. IRF8 Regulates Transcription of Naips for NLRC4 Inflammasome Activation. Cell 2018, 173, 920–933.
- Lesne, S.; Gabriel, C.; Nelson, D.A.; White, E.; Mackenzie, E.T.; Vivien, D.; Buisson, A. Akt-dependent expression of NAIP-1 protects neurons against amyloid- toxicity. J. Biol. Chem. 2005, 280, 24941–24947.
- Kano, O.; Tanaka, K.; Kanno, T.; Iwasaki, Y.; Ikeda, J.E. Neuronal apoptosis inhibitory protein is implicated in amyotrophic lateral sclerosis symptoms. Sci. Rep. 2018, 8, 6.
- Crocker, S.J.; Wigle, N.; Liston, P.; Thompson, C.S.; Lee, C.J.; Xu, D.; Roy, S.; Nicholson, D.W.; Park, D.S.; MacKenzie, A.; et al. NAIP protects the nigrostriatal dopamine pathway in an intrastriatal 6-OHDA rat model of Parkinson’s disease. Eur. J. Neurosci. 2001, 14, 391–400.
- Richter, B.W.; Mir, S.S.; Eiben, L.J.; Lewis, J.; Reffey, S.B.; Frattini, A.; Tian, L.; Frank, S.; Youle, R.J.; Nelson, D.L.; et al. Molecular cloning of ILP-2, a novel member of the inhibitor of apoptosis protein family. Mol. Cell Biol. 2001, 21, 4292–4301.
- Zhu, L.; Zhou, W.; Zhu, X.; Xiang, S.; Wang, S.; Peng, Y.; Lu, B.; Tang, P.; Chen, Q.; Wu, M.; et al. Inhibitor of apoptosis proteinlike protein2: A novel growth accelerator for breast cancer cells. Oncol. Rep. 2018, 40, 2047–2055.

