Survivin is among the most studied members of the IAP family. The protein was discovered in the late nineties as its smallest member involved in fetal development and cancer progression. Survivin is downregulated in most terminally differentiated cells and re-expressed in the majority of solid and liquid human tumors investigated
[55][56][55,56]. Survivin is a prime example of a multifunctional protein involved in a variety of regulatory circuits in tumor cells
[37][57][37,57]. By this, a present conception is that most IAPs, except for XIAP, block apoptosis by mechanisms other than direct caspase inhibition
[58], but via cooperative interactions with other partners. Thus, an association of Survivin with hepatitis B X-interacting protein (HBXIP) and/or XIAP inhibits caspases, while binding to SMAC/Diablo counteracts this activity. Moreover, Survivin is expressed in a cell cycle regulated manner participating in cell division as an interactor of chromosomal passenger complex (CPC) proteins INCENP, Borealin and Aurora-B
[59][60][59,60]. In malignant cells, however, Survivin is regulated independently of mitosis by a variety of oncogenic pathways. Further, Survivin is a predominantly nucleocytoplasmic protein; however, shuttling to or from other compartments like the nucleus is mediated by Exportin-1, irradiation and post-translational regulations such as homodimerization and acetylation of residue K129
[61][62][61,62]. Survivin is subjected to multiple post-translational modifications that mostly are decision-makers on its functions and fate of the host. For instance, phosphorylation of residue T34 by p34(cdc2)-cyclin B1 facilitates proper Survivin-caspase-9 interaction that results in inhibition of apoptosis
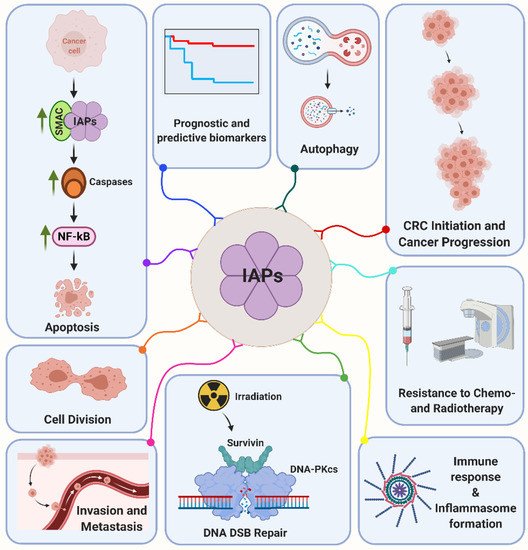
[63]. In addition, Survivin is a radiation-inducible factor mediating the cellular radiation response in a multitude of tumors including colorectal cancer
[64][65][66][64,65,66]. By this, Survivin accumulates in the nucleus and interacts with a prime non-homologous end joining repair factor DNA-dependent protein kinase (DNA-PKcs)
[67][68][67,68]. Survivin forms a heterotetramer complex with DNA-PKcs that results in a conformational change on the DNA-PKcs phosphoinositide 3-kinase domain with enhanced enzymatic activity and detection of differentially abundant phosphopeptides and proteins implicated in the DNA damage response
[69].