Tumor suppressor 53 (p53) is a multifunctional protein that regulates cell cycle, DNA repair, apoptosis and metabolic pathways. In colorectal cancer (CRC), mutations of the gene occur in 60% of patients and are associated with a more aggressive tumor phenotype and resistance to anti-cancer therapy. In addition, inhibitor of apoptosis (IAP) proteins are distinguished biomarkers overexpressed in CRC that impact on a diverse set of signaling pathways associated with the regulation of apoptosis/autophagy, cell migration, cell cycle and DNA damage response. As these mechanisms are further firmly controlled by p53, a transcriptional and post-translational regulation of IAPs by p53 is expected to occur in cancer cells. Here, we aim to review the molecular regulatory mechanisms between IAPs and p53 and discuss the therapeutic potential of targeting their interrelationship by multimodal treatment options.
1. Introduction
Colorectal cancer (CRC) accounts for around 10% (more than 1.2 million cases) of annually diagnosed malignancies in the world. It is the fourth most mortal cancer with about 900,000 deaths per year and the incidence is predicted to increase approximately to 2.5 million new cases by 2035 [
1]. CRC development is characterized by a multistep process involving a series of histological and morphological changes triggered by a sequential accumulation of specific genomic alterations [
2]. Adenomatous polyposis coli (APC) gene mutations occurring in normal colon epithelial cells are among the early incidents of a complex tumorigenesis, resulting in abnormally growing benign precancerous polyps (adenomas and sessile serrated polyps) that, over time acquire the ability to invade the bowel wall and trigger low-grade dysplasia. Successively, promoted by Kirsten rat sarcoma virus (KRAS) oncogene activation and serine/threonine-protein kinase B-Raf (BRAF) mutations along with chromosomal (microsatellite) instabilities, high-grade dysplasia will develop that accelerate transformation to malignant progression and invasive carcinoma by further accumulating p53 mutations. Finally, these local malignancies may acquire the potential to metastasize to local lymph nodes and distant organs [
1,
3]. TP53 gene mutation frequency is 60% in colorectal cancers with the vast majority of mutations located in the DNA-binding domain of the protein. About 60% of TP53 mutations result in an abrogated function of one allele (loss of heterozygosity); however, this can have a dominant negative effect (DNE) and repress wild-type (wt)-p53 functions. By contrast, gain of function (GOF) mutations may induce tumor initiation and progression as well as cancer stemness, invasion, migration and therapy resistance [
4,
5,
6].
In dependence of tumor stage, location and lymph node status, CRC is treated by surgery, neoadjuvant (before surgery) or adjuvant chemotherapy (after surgery) with or without concurrent irradiation. By this, treatment of rectal adenocarcinoma represents a particularly good example for a successful implementation of multimodal concepts in cancer management. The establishment of neoadjuvant therapy based on chemoradiation (CRT) prior to surgical resection was a turning point in the treatment of this entity resulting in substantially reduced local recurrence rates and improved survival by the inclusion of oxaliplatin [
7,
8]. However, despite identical tumor histology and comparable tumor stages, patient’s response to neoadjuvant CRT ranges from a clinically and pathologically confirmed complete remission in 10–30% of cases to progression under treatment [
9]. This variable tumor response further displays a strong prognostic impact and significantly correlates with disease-free (DFS) and overall survival (OS) [
10,
11], while a comprehensive understanding of the molecular basis that defines the individual therapy response is still at its early stage. Among the molecular tumor determinants associated with carcinogenesis, enhanced proliferation, invasion, migration and resistance to anticancer treatment, members of the inhibitor of apoptosis (IAP) family proteins, most pronounced cellular IAP (cIAP1, cIAP2), X chromosome linked IAP (XIAP) and Survivin, have gained increasing interest [
12]. In this review, we aim to illustrate a mechanistic interrelationship between IAPs and p53 that may pave the way to develop new combinational therapies to overcome mutant p53 and IAPs based therapy resistance in CRC.
2. Biology and Functions of p53, a Brief Introduction
The p53 protein and poly(ADP-ribose) polymerases (PARPs) are considered to be the “guardians of the genome” due to their role in conserving genetic stability by preventing mutations and mediating tumor suppression via a tightly regulated network in response to stress signals, which results in either cell death or survival [
4]. PARP-1, by a direct poly(ADP-ribosyl)ation of the p53 protein results in a nuclear accumulation and transcriptional activation of p21 [
13] or is required for ATM-mediated p53 activation and gene expression [
14]. In addition, epigenetic repression (p53) and activation (PARP-1) of DNA (cytosine-5)-methyltransferase 1 (DNMT1) activity, a key enzyme implicated in the silencing of DNA repair genes, may represent an indirect interrelationship between both proteins [
15].
The major structural part of the p53 protein covers a central DNA-binding domain (DBD), which is connected to the tetramerization domain by a linker region. The regulatory domain is located adjacent to the homo-oligomerization (OD) domain at the protein’s carboxy-terminal end. The vast majority of p53 mutations are located in the DNA-binding region [
16]. Transcriptional functions of p53 are mediated by binding to variable consensus sequences in responsive elements in the promoter of target genes. Moreover, p53 also regulates genes partially or completely lacking these consensus sequences dependent on their secondary structure [
17]. In addition, p53 directly binds and regulates proteins such as ataxia telangiectasia mutated (ATM) kinase and transcription factors such as Y-box-binding protein (YB-1) [
18]. This diversity enables multiple regulatory functions in cellular pathways but needs to be tightly controlled. In that context, a negative feedback loop via murine double minute 2 homologue (MDM2) and MDM4 controls p53-mediated transcriptional and post-transcriptional activity and keeps p53 at low levels under physiological conditions [
19,
20]. Following DNA damage, p53 is activated by post-translational modifications, e.g., phosphorylation by phosphatidylinositol 3-kinase-related kinase (PIKK)-family members ATM, ataxia telangiectasia and Rad3-related (ATR) and DNA-dependent protein kinase catalytic subunit (DNA-PKcs) or indirect phosphorylation by ATM/ATR/DNA-PKcs substrates checkpoint kinases 1 (CHK1) and CHK2 [
19,
21]. These modifications result in p53 stabilization, activation and nuclear translocation, followed by p53-mediated transcription of a plethora of target genes, involved in cell cycle regulation, DNA damage repair and apoptosis [
22].
3. Structure and Function of the Inhibitor of Apoptosis Protein Family (IAP)
The IAP family was first described in 1993 as a class of baculoviral proteins characterized by a functional baculovirus IAP repeat (BIR) domain [
23] that prevented apoptosis of insect cells during viral infection [
24]. Since their discovery, BIR containing (BIRC) proteins were reported in yeast, insects and mammalians. The human IAP family currently covers eight members, including neuronal apoptosis inhibitory protein (NAIP/BIRC1), cellular IAP1 (cIAP1/BIRC2), cellular IAP2 (cIAP2/BIRC3), X-chromosome-linked IAP (XIAP/BIRC4), Survivin (BIRC5), BIR repeat-containing ubiquitin-conjugating enzyme (BRUCE/Apollon/BIRC6), LIVIN (BIRC7) and human IAP-like 2 (hILP2/BIRC8) [
25,
26]. The family members differ substantially in protein size and functional domains but share at least one of the family-defining BIR domain facilitating protein-protein interactions with other factors. Additional functional domains include a centrally located ubiquitin associated (UBA) domain present in cIAP1 cIAP2, XIAP and hILP2 to allow these proteins to bind to poly-ubiquitin chains, or a carboxy-terminal localized really interesting new gene (RING) domain conferring ubiquitin ligase activity and mediating signal transduction, protein-protein interactions, and transcription [
25]. Further, a caspase activation and recruitment domain (CARD), unique to cIAP1/2, helps to control their ubiquitin ligase activity and stability. In addition, NAIP includes a NAIP-C2TA-HETE-TEP1 nucleotide-binding and oligomerization domain (NACHT) which functions in apoptosis inhibition and major histocompatibility complex (MHC) class II transcriptional activation and a leucine-rich repeat (LRR) domain with a role in signaling pathways of innate immunity and host-pathogen recognition [
27]. Finally, Survivin carries an amphipathic α-helical coiled-coil domain at the C-terminus, common in microtubule-associated proteins [
28].
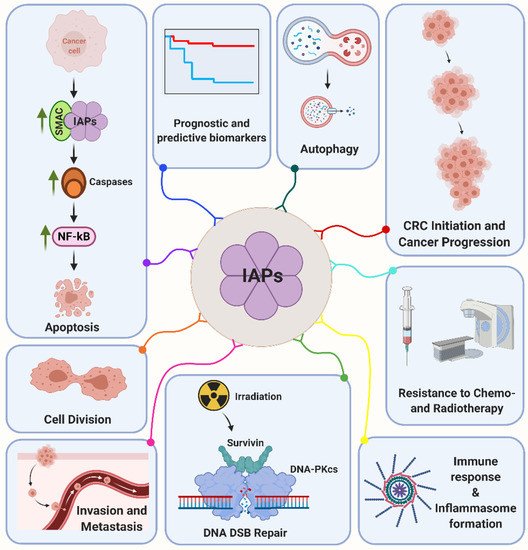
Although IAPs are primarily considered as sole inhibitors of apoptosis, growing evidence evolves regarding their vital impact as transduction intermediates in a diverse set of signaling pathways associated with the regulation of cell migration, cell cycle and DNA damage response. Some of these functions will briefly be described below and are summarized in .
Figure 1. Inhibitor of apoptosis proteins (IAPs) are multifunctional proteins that regulate a variety of key cellular mechanisms such as apoptosis, cell division, invasion and metastasis, autophagy, DNA double-strand break (DSB) repair, cancer progression, immune response and inflammasome formation. Moreover, IAPs are associated with radiation and chemotherapy resistance and are considered to be valuable prognostic and predictive biomarkers in colorectal cancer (CRC). Please see the text for a more detailed discussion. Abbreviations: DNA-PKcs, DNA-dependent protein kinase, catalytic subunit; NF-κB, nuclear factor kappa B; SMAC, second mitochondrial activator of caspases.
3.1. cIAP1 and cIAP2
Primarily, cIAP1 was discovered by its involvement in inflammation/apoptosis signaling interacting with tumor necrosis factor receptor-2 associated factors (TNFR2-TRAFs) via its N-terminal BIR domain [
29]. The major biological activities of cIAP1 cover a positive regulation of the canonical transcription factor nuclear factor kappaB (NF-κB) activation pathways. By this, cIAP1 complexes with TRAF2, Src homology 2 domain-containing protein tyrosine phosphatase 1 (SHP1), Src and myeloid differentiation primary response 88 (MyD88) to promote canonical activation of NF-κB [
30]. Briefly, in the TNFR1 complex, cIAP1/2 serve as ubiquitin ligases for receptor-interacting serine/threonine-protein kinase 1 (RIPK1), which is needed for TNF-α mediated NF-κB and mitogen-activated protein kinase (MAPK) signaling, gene expression, differentiation, mitosis and inhibition of both, caspase-dependent and -independent cell death. In addition, cIAP1/2 limit the non-canonical NF-κB activation pathway. Here, cIAP1/2 act as ubiquitin ligases that target NF-κB-inducing kinase (NIK) for degradation. Further, upon viral infection, cIAP1/2 ubiquitinate TRAF3/6 which is an essential factor for NF-κB deregulation, while attenuation of cIAP1/2 impedes an antiviral response via inhibition of the virus-triggered activation of NF-κB, interferon regulatory factor 3 (IRF3) and interferon-beta (IFN-β) induction [
31].
The expression level of cIAP1 is regulated by a variety of transcriptional and post-translational mechanisms including microRNAs (miRNAs) and proteasomal degradation. For instance, microRNA-29c (miR-29c) binds to the 3′-UTR of cIAP1 effectively downregulating its mRNA and protein levels [
32]. Further, downregulation of ubiquitin thioesterase OTU domain ubiquitin aldehyde binding 1 (OTUB1) enhances the degradation of cIAP1 and inhibits the TNF-related weak inducer of apoptosis (TWEAK)-induced MAPK and NF-κB pathway [
33]. Upon genotoxic stress, a bilateral cell death regulation involving the ripoptosome (RIP1/3-FADD-Caspase-8-c-FLIP) complex is reported: cIAP1 and cIAP2 directly ubiquitinate RIP1 which associates with the pro-survival transforming growth factor beta-activated kinase 1 (TAK1) and triggers proteolytic degradation of the ripoptosome complex while attenuation of IAPs allows the proper formation of the complex [
34].
Beside its function in concert with cIAP1, cIAP2 is involved in response to metal stress, DNA repair and together with XIAP and Survivin in exosomal secretion [
35,
36,
37]. The latter may not only serve as warning signals, but may also play a role in providing protection to the cancer cells against potential dangers in the tumor microenvironment [
38].
As a response to histone deacetylase (HDAC) inhibitor Panobinostat treatment, cIAP2 exhibits the highest upregulation in line with a decreased level of DNA double-strand break (DSB) repair protein meiotic recombination 11 homolog (MRE11). Moreover, cIAP2 directly interacts with MRE11, promotes its ubiquitination and directs it to degradation that in turn delays DNA DSB repair resulting in increased radiation sensitivity [
35].
3.2. XIAP
Human XIAP was initially discovered as an IAP-like apoptosis inhibitor protein by its homology to baculovirus IAP genes [
39]. XIAP is an archetypical IAP protein that, in contrast to other family members, inhibits the active catalytic sites of caspases-3 and caspases-7 in a direct manner and interferes with the dimerization and activation of caspase-9. This prevents their downstream effector functions, including the release of mitochondrial IAP antagonists such as second mitochondrial activator of caspases (SMAC/Diablo) and the serine peptidase HtrA2/Omi, that bind XIAP’s BIR domains, releasing active caspases into the cytosol [
40]. In addition, recent studies presented XIAP as a multifunctional protein involved in cellular and metabolic regulatory circuits such as invasion, migration, necroptosis, oxidative stress, inflammasome formation and autophagy [
41,
42,
43,
44,
45,
46,
47,
48,
49,
50]. XIAP´s BIR domain 1 mediates activation of stress-responsive signaling pathways, such as Jun Kinase (JNK) [
51] and MAPK phosphorylation cascade that in turn activate NF-κB. XIAP also activates NF-κB by promoting the translocation of the p65 subunit to the nucleus and by degradation of the NF-κB inhibitor IκB [
52]. XIAP empowers interleukin-17 mediated NF-κB activation and caspase-3 inhibition that drives colon tumor formation [
44]. In line with Survivin, XIAP represents a radiation resistance factor and attenuation of the protein triggers radiation response in CRC cancer cell lines [
53,
54].
3.3. Survivin
Survivin is among the most studied members of the IAP family. The protein was discovered in the late nineties as its smallest member involved in fetal development and cancer progression. Survivin is downregulated in most terminally differentiated cells and re-expressed in the majority of solid and liquid human tumors investigated [
55,
56]. Survivin is a prime example of a multifunctional protein involved in a variety of regulatory circuits in tumor cells [
37,
57]. By this, a present conception is that most IAPs, except for XIAP, block apoptosis by mechanisms other than direct caspase inhibition [
58], but via cooperative interactions with other partners. Thus, an association of Survivin with hepatitis B X-interacting protein (HBXIP) and/or XIAP inhibits caspases, while binding to SMAC/Diablo counteracts this activity. Moreover, Survivin is expressed in a cell cycle regulated manner participating in cell division as an interactor of chromosomal passenger complex (CPC) proteins INCENP, Borealin and Aurora-B [
59,
60]. In malignant cells, however, Survivin is regulated independently of mitosis by a variety of oncogenic pathways. Further, Survivin is a predominantly nucleocytoplasmic protein; however, shuttling to or from other compartments like the nucleus is mediated by Exportin-1, irradiation and post-translational regulations such as homodimerization and acetylation of residue K129 [
61,
62]. Survivin is subjected to multiple post-translational modifications that mostly are decision-makers on its functions and fate of the host. For instance, phosphorylation of residue T34 by p34(cdc2)-cyclin B1 facilitates proper Survivin-caspase-9 interaction that results in inhibition of apoptosis [
63]. In addition, Survivin is a radiation-inducible factor mediating the cellular radiation response in a multitude of tumors including colorectal cancer [
64,
65,
66]. By this, Survivin accumulates in the nucleus and interacts with a prime non-homologous end joining repair factor DNA-dependent protein kinase (DNA-PKcs) [
67,
68]. Survivin forms a heterotetramer complex with DNA-PKcs that results in a conformational change on the DNA-PKcs phosphoinositide 3-kinase domain with enhanced enzymatic activity and detection of differentially abundant phosphopeptides and proteins implicated in the DNA damage response [
69].
3.4. BRUCE/Apollon
With a molecular weight of 528 kDa, BRUCE/Apollon is a huge E3 ubiquitin transferase whose mutation causes embryonic lethality [
70,
71]. In concert with other IAPs, one of the main functions of BRUCE is inhibition of apoptosis [
70]. BRUCE binds and ubiquitinates SMAC/Diablo, caspase-9 and mitochondrial serine peptidase HtrA2/Omi to prevent apoptosis by facilitating their proteasomal degradation [
72,
73]. Early in mitosis, BRUCE binds the anaphase-promoting complex/cyclosome (APC/C) and enables the degradation of Cyclin A by ubiquitination independent of cyclin-dependent kinases (CDKs) [
74]. However, in the late phase of cytokinesis, BRUCE is an essential component of the midbody ring and the tubular recycling system under the regulation of mitotic kinesin-like protein-1 to regulate cytokinetic abscission [
75]. Upon DNA damage, BRUCE acts as a scaffold to form a complex with ubiquitin-specific peptidase 8 (USP8) and breast cancer susceptibility gene C terminus-repeat inhibitor of human telomerase repeat transcriptase expression-1 (BRIT1) at the DSBs, which is vital for the formation of BRIT1 DNA damage foci [
76]. BRUCE further regulates ATR-directed signaling pathways in DNA replication stress via interaction with pre-mRNA-processing factor 19, while depletion of BRUCE causes a stalled DNA replication, prevents the activation of ATR and inhibits the phosphorylation of CHK1 and replication protein A [
77].
3.5. LIVIN
LIVIN (37/39 kDa) plays a key role in a multitude of cellular mechanisms and stress responses including radiation response, invasion, hypoxia-resistance and autophagy [
78,
79,
80,
81,
82]. LIVIN inhibits apoptosis by binding both, caspase-3/7 as well as TNFα-induced DEVD-like caspase. In addition, LIVIN indirectly inhibits caspase-9 via apoptotic protease activating factor-1 (Apaf-1) [
83]. However, reports on a bidirectional regulation between caspases and LIVIN revealed that the truncation of LIVIN (28/30 kDa) by caspase-3/7 transforms it to a pro-apoptotic protein [
84,
85]. Chemosensitivity studies in colon cancer cells further revealed LIVIN as a drug resistance gene against etoposide (VP-16) and 5-fluorouracil (5-FU) [
86], while attenuation of the protein significantly decreases the size of colon cancer xenograft tumors [
86,
87]. Upon irradiation, LIVIN overexpression is associated with cellular radioresistance, whereas attenuation of LIVIN decreases radiation-induced cell invasion ability and enhances radiation response [
78,
79].
3.6. NAIP and hILP-2
NAIP was first discovered as spinal muscular atrophy (SMA) related gene whose deletion or mutation was restricted to SMA patients [
88]. However, comprehensive studies over the last years indicate the relevance of NAIP in a variety of different molecular mechanisms and diseases such as cytokinesis, inflammasome formation, amyloid-β toxicity, amyotrophic lateral sclerosis (ALS) and Parkinson’s disease [
89,
90,
91,
92,
93]. The least-studied member of IAP family, hILP-2 is discovered as a protein owing a high sequence homology to XIAP but no inhibitory effect on TNF-mediated apoptosis. Nevertheless, it can inhibit Bcl-2-associated X protein (Bax) or caspase-9 and Apaf-1 triggered apoptosis. Moreover, hILP-2 directly interacts with cleaved caspase-9 [
94] and attenuation of hILP-2 triggers apoptosis and inhibits migration [
95].
This entry is adapted from the peer-reviewed paper 10.3390/cancers13040624