Studies concerning the role of arachidonic acid (AA) and its metabolites in kidney disease are scarce, and this applies in particular to idiopathic nephrotic syndrome (INS). INS is one of the most frequent glomerular diseases in childhood; it is characterized by T-lymphocyte dysfunction, alterations of pro- and anti-coagulant factor levels, and increased platelet count and aggregation, leading to thrombophilia. AA and its metabolites are involved in several biological processes. Herein, we describe the main fields where they may play a significant role, particularly as it pertains to their effects on the kidney and the mechanisms underlying INS. AA and its metabolites influence cell membrane fluidity and permeability, modulate platelet activity and coagulation, regulate lymphocyte activity and inflammation, preserve the permeability of the glomerular barrier, influence podocyte physiology, and play a role in renal fibrosis.
1. Introduction
Idiopathic nephrotic syndrome (INS) is one of the most frequent glomerular diseases in childhood
[1]. It is characterized by proteinuria, caused by podocyte damage, hypoalbuminemia, hyperlipidemia, and edema
[1]. While the exact cause of podocyte damage is still not completely understood
[1], it is well known that hyperlipidemia is related to urinary loss of transport proteins, which carry free cholesterol, and to the consequent compensatory increase in the synthesis of proteins involved in triglyceride metabolism
[1]. Two theories have been proposed to explain the pathogenesis of edema in INS. According to the classical underfill hypothesis, hypoalbuminemia reduces plasma oncotic pressure, which leads to sodium and water retention and water leakage into the interstitium
[2]. Meanwhile, the overfill hypothesis postulates proteinuria to be the primary cause of sodium retention, with consequent volume expansion and leakage of excess fluid into the interstitium
[3].
Other biochemical alterations were also described in INS, such as changes in pro- and anti-coagulation factors’ levels and increased platelet count and aggregation, leading to a hypercoagulable state
[4].
Based on their response to corticosteroid therapy, children with INS are classified as steroid-sensitive patients, which includes those with infrequent relapses, frequently relapsing or steroid-dependent patients who present a favorable prognosis, or steroid-resistant patients, who carry an unfavorable prognosis in the majority of cases. Histopathology usually reveals minimal change of disease, which is characterized by normal glomerular appearance on light microscopy and evidence of podocyte foot processes’ alterations on electron microscopy; focal segmental glomerulosclerosis and interstitial fibrosis may be found in steroid-resistant cases
[5][6].
The pathogenesis of INS has not yet been fully clarified. Excluding genetic causes, the main theory for immune-mediated cases involves a dysfunction of T lymphocytes, which would switch to the production of still poorly defined permeability factors that interfere with the expression and/or function of key proteins in the podocyte, thus being the main culprits of proteinuria
[7]. Candidates for the circulating factors that affect glomerular permeability include angiopoietin-like 4 (ANGPTL4), cardiotrophin-like cytokine-1 (CLC-1), and soluble urokinase plasminogen activator receptor (suPAR)
[1].
Arachidonic acid (AA) is a long-chain polyunsaturated fatty acid of the omega-6 group and represents 7% to 10% of total circulating fatty acids; it is the second most abundant omega-6 fatty acid in the human body
[8] (), with linoleic acid (LA) being the first. AA is synthesized endogenously from LA through three steps mediated by two enzymes, desaturase and elongase, and may also be derived from the diet. In turn, AA is a substrate of elongases for the synthesis of longer fatty acids of the omega-6 series.
Table 1. Blood omega-6 levels in healthy subjects. Linoleic acid, AA, omega 6, total saturated fatty acid, monounsaturated fatty acids, and total omega-3 levels in human subjects. AA is the second highest fatty acid of the omega-6 series. Data are expressed as percentage of total fatty acids
[4].
| |
Neonates |
Children |
Adults |
Elderly |
| Linoleic acid (%) |
4.61 ± 1.06 |
17.67 ± 1.92 |
18.41 ± 2.87 |
17.64 ± 2.89 |
| Arachidonic acid (%) |
13.14 ± 1.73 |
8.33 ± 1.04 |
8.51 ± 1.38 |
8.32 ± 1.40 |
| Total omega 6 (%) |
22.99 ± 2.13 |
28.97 ± 2.19 |
29.79 ± 3.13 |
28.78 ± 3.24 |
| Total saturated fatty acids (%) |
46.10 ± 3.16 |
44.32 ± 1.61 |
39.47 ± 2.3 |
39.83 ± 2.16 |
| Total monounsaturated fatty acids (%) |
26.15 ± 2.76 |
24.39 ± 2.07 |
27.20 ± 3.08 |
27.83 ± 3.27 |
| Total omega 3 (%) |
4.76 ± 0.89 |
2.31 ± 0.50 |
3.54 ± 1.05 |
3.55 ± 0.95 |
AA is metabolized by three types of oxygenases: cyclooxygenase (COX), lipoxygenase (LOX), and cytochrome P450, leading to the generation of eicosanoids, namely prostaglandins, thromboxane, leukotrienes, and hydroxyeicosatetraenoic acids.
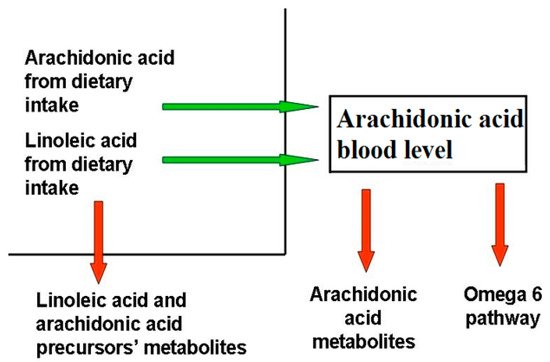
Blood AA levels do not reflect its synthesis and metabolization pathways (), as they are maintained as constant, even at the expense of other biological factors, as observed in patients with epidermolysis bullosa
[9], where, despite the large amount of active AA metabolites, the AA level is comparable to that of healthy controls. This phenomenon has been observed in several other chronic inflammatory disorders, for instance cystic fibrosis
[10], even if the exact mechanism behind it is unclear.
Figure 1. Factors determining blood arachidonic acid levels. The factors influencing AA levels belong to two categories, those that increase (green arrow) AA blood levels, like diet uptake and the omega-6 pathway from LA, and those that decrease them (red arrow), like the synthesis of omega-6 FA downstream AA, AA metabolization, and the reduction of AA precursors due to their metabolization. Diseases able to modify AA blood levels act through the same mechanisms.
AA is involved in several biological processes, either in health or disease. Herein, we describe its role in nephrotic syndrome from a biological and clinical perspective. AA influences cell membrane fluidity and permeability and modulates platelet function and immune system activation; furthermore, it affects glomerular and tubular function, the physiopathology of podocyte, and the process of renal fibrosis. We also detail the interactions between AA and the common drugs prescribed for INS treatment. Finally, the role of dietary AA balance and its nutritional sources are discussed.
2. Cell Membrane Fluidity and Permeability
It was recently described that erythrocyte membranes of patients with INS differ from those of normal subjects, particularly due to reduced membrane fluidity
[11].
AA is one of the most abundant fatty acids in the cell membrane, to which it endows mobility and flexibility
[12][13]. The fatty acid composition determines the viscosity of the cell lipid bilayer and membrane fluidity, thus directly affecting the function of specific membrane proteins, like, for example, those involved in cellular inflammatory signaling, namely lymphocyte function-associated antigen 1 (LFA-1), intercellular adhesion molecule 1 (ICAM-1), and cluster of differentiation 2 (CD2)
[12][13].
With regard to membrane permeability, AA acts on Ca
2+ cell load
[10] with a double effect: at low micromolar concentrations it increases Ca
2+-ATPase activity, while at higher concentrations it reduces ATPase activity. This may be due to an unspecific and non-physiological inhibitory effect on the hydrolytic activity of P-type ATPase. ATPases are a superfamily of lipid pumps involved, among other functions, in secretion and absorption at the kidney level; these pumps are blocked by protein kinase C inhibitors
[14]. AA increases membrane permeability to calcium, which is a key factor for platelet activation
[15].
AA may act on ion channels by either binding to or inserting among the membrane molecules, thus modifying the mechanical properties of the cell membrane and modulating channel function
[16].
AA also has a direct effect on several membrane potassium channels, either by accelerating their inactivation (in particular, the A-type channels and delayed rectifier channels), or by inducing the activation of large-conductance voltage-independent channels. The two-pore domain potassium channels are inactivated by AA as well, in contrast to what usually occurs with classical K channel-blocking drugs
[16]. Transient receptor potential channels (TPR) are instead activated directly by AA and its lipoxygenase (LOX)-derived metabolites
[16] (namely, 12- and 15-(
S)-hydroperoxyeicosatetraenoic acids, 5- and 15-(
S)-hydroxyeicosatetraenoic acids, and leukotriene B
4). LOX metabolites can activate the TPR channel by virtue of their structure that mimics the capsaicin structure
[17]. Interestingly, AA and its metabolic byproducts effects on calcium and potassium balance at the membrane level have been hypothesized to underlie the molecular-related derangements in INS
[6].
As it concerns membrane fluidity, albumin is the main fatty acid-binding protein in extracellular fluid, having seven fatty acid-binding sites
[18]. Albumin increases AA release from cell membranes in a concentration-dependent manner, by interacting with membrane phospholipids on the extracellular surface; in particular, positively charged arginine residues at or near albumin’s binding sites for LCFA interact with AA, determining its release from the phospholipid layer
[19]. Thus, albumin decreases cell membrane permeability of endothelial and circulating cells to water and small solutes
[19].
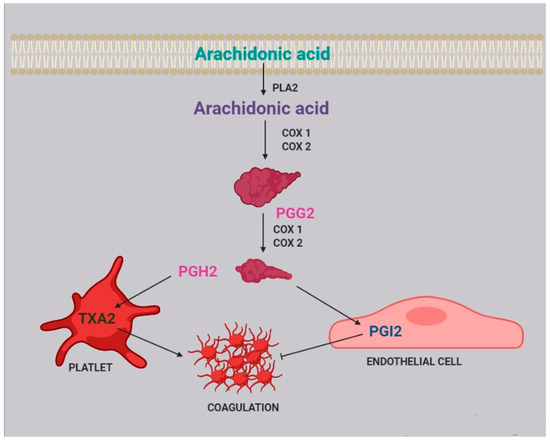
3. Platelet Aggregation and Coagulation
Two of the most active compounds related to platelet function are thromboxane and prostacyclin, both metabolites of AA
[20]. AA is released from platelet membrane by phospholipase A
2 (PLA
2), which hydrolyzes the bond between the second fatty acid of phospholipids and the glycerol molecule. The released AA is then metabolized by cyclooxygenase
[21], generating prostaglandin G
2, and thereafter prostaglandin H
2. Afterwards, two different pathways can take place: the first one, within the platelets, leads to the synthesis of thromboxane A
2 (TXA
2) and subsequently B
2 (TXB
2); the second one, within the endothelial cells, leads to the synthesis of prostacyclin (PGI
2) ().
Figure 2. Role of arachidonic acid in the coagulation process. Arachidonic acid is released from cell membranes by phospholipase A2 (PLA2) and subsequently metabolized by COX1 and COX2 to obtain prostaglandin G2 (PGG2) and H2 (PGH2).
TXA
2 stimulates platelet activation and aggregation, via platelet fibrinogen-binding αIIbβ
3 receptors
[4]. Prostacyclins, in contrast, inhibit platelet activation by activating G protein-coupled receptors on platelets and endothelial cells. Upon binding to the prostacyclin receptor, PGI
2 induces adenyl cyclase cAMP production, which in turn inhibits platelet activation
[22].
A higher incidence of increased platelet aggregation and thromboembolism has been reported in nephrotic syndrome, in relation to consistently elevated levels of fibrinogen. Moreover, both hyperlipidemia and hypoalbuminemia, which are characteristic findings of nephrotic syndrome, increase thromboxane availability, through the production of TXA
2 precursors and the removal of TXA
2 inhibitors
[23]. The exact mechanism underlying this process is still unknown, but it probably involves an increase in PLA
2 activity, related to the abnormally high cholesterol levels
[24]. Therefore, arachidonic acid, which is the precursor of thromboxane, may be considered a crucial player in the platelet-related coagulation process.
4. Immune System
The therapeutic efficacy of Rituximab in modifying the course of steroid-dependent nephrotic syndrome suggested that B cells play a key role in the pathogenesis of INS. This was recently confirmed by evidence of a pathological increase in memory B cells in INS
[25]. Moreover, other studies showed a decrease of Treg cells
[26], dysregulation of T-cells
[27], lower levels of NK and NKT cells, and increased levels of inflammatory markers during proteinuria
[28][29]. These studies confirm
[1] that the immune system plays a pivotal role in non-genetic INS and specifically in the loss of the glomerular barrier function, by activating the inflammatory process against podocytes.
In immune cells, like lymphocytes, neutrophils, and monocytes, AA constitutes about 20% of total fatty acids, while EPA and DHA constitute 1% and 2.5%, respectively
[30]. It was reported that oral administration of omega-3 fatty acids changes the pattern of production of eicosanoids, by increasing resolvins production, thus affecting phagocytosis, T-cell signaling, and antigen presentation capability. These effects seem to be mediated at the membrane level
[30].
The distribution of AA within intracellular lipid pools in inflammatory cells has an important role in regulating eicosanoids production. In fact, a pool of AA was identified within the triglycerides of mast cells, eosinophils, monocytes, and platelets
[31].
When inflammatory cells are activated, AA is released from membrane phospholipids into the cell and partially incorporated into intracellular triglycerides, ready to supply membrane phospholipids again after cell activation has ended
[32].
Thus, AA metabolites can act in several ways on lymphocyte activity, affecting inflammation levels
[32][33][34] () and possibly the course of INS.
Table 2. Effects of AA metabolites on immune cells
[35]; AA metabolites affect immune cells in various ways, modulating the immune response and inflammation.
| Cell Type |
AA Metabolite |
Effect |
| Basophil |
PGD2 |
Stimulates basophil chemotaxis |
| Eosinophil |
PGD2 |
Stimulates eosinophil chemotaxis |
| Blocks eosinophil apoptosis |
| Activates eosinophils |
| Naive t cell |
TXA2 |
Inhibits proliferation of naive T cells |
| B-cell |
PGE2 |
Enhances IgE class switching by B cells |
| Dendritic cell |
LTB4 |
Stimulates DC production of IL-6 |
| LTC4 |
Participates in cell migration |
| Enhances cells activation and functions |
| PGD2 |
Inhibits cells migration |
| PGE2 |
Stimulates IL-10 production |
| Modulates cell migration |
| Downregulates major histocompatibility complex C class II expression |
| Inhibits IL-12 and IFN-ã production |
| Inhibits the expression of CCL3/CCL4 |
| PGJ2 |
Induces apoptosis |
| TXA2 |
Inhibits interaction with T cell |
| Langerhans cell |
PGE2 |
Promotes the migration and maturation of Langerhans cells |
| PGD2 |
Inhibits cells migration |
| Lymphocyte |
PGE2 |
Inhibits interactions with endothelial cell |
| Macrophage |
PGE2 |
Suppresses cytokine production |
| Suppresses chemokine expression |
| PGJ2 |
Inhibits release of IL-10 and IL-12 |
| Mast cell |
PGE2 |
Enhances antigen-stimulated degranulation |
| Neutrophil |
LTB4 |
Activates cells |
| NK cell |
PGE2 |
Inhibits IL-12 and IFN-ã production |
| T cell |
LTB4 |
Enhances cell recruitment |
| PGE2 |
Inhibits cell proliferation |
| TXA2 |
Inhibits interactions with dendritic cells |
| Regulates the elimination of self-reactive cells |
| Increases cell proliferation and activation |
| Enhances local cytotoxic cell function |
| Th2 |
PGD2 |
Stimulates chemotaxis |
Regarding B, NK, and T cells, the main AA metabolites involved are PGE2, LTB4, and TXA2.
PGE
2 is produced by nearly all cells within the body
[36]. Secreted PGE
2 acts in an autocrine or paracrine manner through its four cognate G protein-coupled receptors EP1 to EP4
[37]. It inhibits T-cell and NK-cell proliferation, as well as IFN-γ and IL-12 production
[38], binding their cell-surface receptors
[35]. PGE
2 also inhibits B-cell activation secondary to IL-4 stimulation in a specific manner and enhances IgE and IgG1 production
[39].
LTB
4 exerts pleiotropic effects on lymphocytes and regulates the immune response in a dynamic, cell type- and context-dependent manner: LTB
4 enhances T-cell recruitment, it inhibits de novo iTreg generation and increases interleukin-17 (IL-17) cytokine production during T-cell differentiation. LTB
4 also regulates the migration of various lymphoid-derived cell types in different ways that vary depending on disease and tissue.
[40].
TXA
2, another product of AA metabolism, inhibits naïve T-cell proliferation and exerts several effects on mature T lymphocytes: it inhibits T-cell interaction with dendritic cells, increases T-cell proliferation and activation, and has been shown to topically enhance the cytotoxic activity of immune cells
[38].
Moreover, eosinophils, mast cells, macrophages, dendritic cells, and Th2 lymphocytes have surface membrane receptors for arachidonic-derived metabolites, in particular for prostaglandin D
2, cysteinyl leukotrienes D
4 and E
4, and lipoxin A
4 [33], but these findings have not been confirmed so far in patients with INS.
A pharmacological modulation of AA metabolites could decrease the inflammatory damage to the podocyte. The pathogenetic role of AA is supported by the fact that medications have been recently administered to target AA metabolism and decrease kidney inflammation
[21][34]. They include aspirin, nimesulide, licofelone, baicalein, and others. Some of them are in the early stages of development for kidney diseases like diabetic nephropathy, glomerulonephritis, and idiopathic membranous nephropathy
[34].
5. Kidney Glomerular and Tubular Function
Epoxyeicosatrienoic acids (EETs) are produced in several tissues, like the heart, the muscles, the kidneys, the pancreas, the lungs, and the brain
[41], but mainly in the vascular endothelium, in response to various PLA
2-activating stimuli, EETs activity could be reduced by metabolization made by soluble epoxyde hydrolase (sHE)
[42].
EETs modulate kidney function acting directly on tubular ionic transport, vascular tone, and cellular proliferation, and have a nephro-protective role
[43] due to their anti-inflammatory properties.
EETs in fact induce vasodilatation in an autocrine manner
[44] and have anti-apoptotic activity; it was also reported that their generation is reduced in case of renal disease
[42], even if no explanation regarding such a mechanism was given.
Glomerular inflammation is mitigated by EETs, which decrease the influx of neutrophils and macrophages and decrease the production of cytokines, monocyte chemotactic protein-1, TNF-α, macrophage inflammatory protein 2, and ICAM-1
[45]. The protective effect is due to EETs renal vasodilator and antipressor response to salt loading, through the inhibition of renal tubular Na
+ reabsorption and the increase of Na
+ renal excretion, resulting in an anti-hypertensive effect
[46] that is probably mediated by A2A receptors
[47].
20-HETE, another eicosanoid derived from AA metabolism, shares the same protective properties as EETs
[48]. It plays a predominant role in the regulation of renal tubular and vascular function, and variants in the genes encoding for the enzymes that produce 20-HETE are associated with hypertension
[49].
It has been shown that sustained production of 20-HETE in the glomerulus is required to maintain the glomerular permeability barrier to albumin
[48]. It is still unclear which cell types in the glomerulus express the CYP enzymes that synthetize 20-HETE, and the exact mechanisms by which this molecule influences the glomerular permeability barrier are yet to be defined as well
[48], although it is likely that its effects are mediated by the modulation of Na
+-K
+-ATPase, Na
+-K
+- 2Cl
− cotransporter, and K
+ channel activity in nephrons
[49] through the activation of the PKC pathway
[50].
20-HETE is also involved in podocyte apoptosis, by regulating the canonical transient receptor potential-6 (TRPC6) channels and increasing the Ca
2+ flux
[51].
In patients affected by nephrotic syndrome who develop early hypertension
[52], a decreased concentration of 20-HETE in the proximal tubule has been observed
[53] in association with increased albumin permeability in the glomeruli, which worsens proteinuria and glomerular injury
[54]; this finding supports the role of 20-HETE in preserving glomerular permeability barrier to albumin. However, it is unknown if the reduction of 20-HETE causes or is caused by hypertension.
In conclusion, 20-HETE may act in different ways (protective or pro-apoptotic) in different cell types and kidney regions.
6. Podocyte Physiopathology and Infections
It is well known that, in the course of nephrotic syndrome, infections lead to an exacerbation of proteinuria
[55] and are an important risk factor for relapses
[1][56][57].
Infections activate the immune system, triggering the inflammatory cascade. It was recently reported that during inflammation two enzymes are induced: 15-lipoxygenase (15-LO) and secreted phospholipase A
2 (sPLA
2)
[58]. Notably, 15-LO is expressed in human podocytes
[59], while sPLA
2 is expressed in platelets, neutrophils, eosinophils, and macrophages
[60]. sPLA2 releases AA from membrane phospholipids
[51][61] acting in a paracrine way.
In glomerular podocytes, intracellular free AA is metabolized to PGE
2, which, by interacting with the EP 4 receptor (prostaglandin E
2 receptor 4) expressed by podocytes, reduces AA release
[52]. This loop regulates podocyte function in both physiological and pathological conditions and is able to change PGE
2 synthesis
[62].
As described above, during the course of an infection, intracellular AA levels increase due to the action of sPLA
2. It was reported that in podocytes, an excess of AA activates protein kinase A, which in turn promotes c-Abl activation and nephrin phosphorylation, thus causing actin cytoskeleton remodeling and podocyte injury
[63]. This mechanism could partially explain the frequent recurrence of proteinuria during infectious episodes in children. Moreover, an increase in sPLA
2 1B levels and PLA
2R expression has been observed to be positively associated to podocyte apoptosis in kidneys of patients with idiopathic membranous nephropathy
[64].
Podocyte foot process injury and podocyte apoptosis due to cytoskeleton remodeling were also attributed to a change of Ca
2+ efflux
[51], driven by 20-HETE, the main AA metabolite. It has also been observed that 20-HETE increases the current Ca
2+ flowing through TRPC6 channels in the podocyte
[51], which are located at the slit diaphragm, possibly leading to cellular injury.
7. Renal Fibrosis
Renal fibrosis is a process that progresses independently of the primary renal disease
[65] and represents a failed wound-healing process of the kidney tissue. Renal biopsies of patients with steroid-resistant nephrotic syndrome often show glomerulosclerosis and interstitial fibrosis, which are associated with progression to end-stage kidney disease in more than 50% of cases
[65], a poor prognosis that heightens the necessity of increasing our knowledge of the mechanisms underlying fibrosis.
Renal fibrosis is characterized by connective tissue deposition in the kidney parenchyma, particularly in the interstitial space and within the walls of glomerular capillaries, and by the consequent cellular processes. Fibrosis also interferes with normal tubular function, leading progressively to organ failure
[65][66].
The scar tissue contains fibrillar collagen I and III as well as some constituents of the normal capillary basement membrane, like collagen IV and V, fibronectin, laminin, perlecan, and heparin
[66].
Fibrosis is associated with leukocyte recruitment, angiogenesis, vascular leak, and the appearance of myofibroblasts. In particular, both the glomerulus and the interstitium attract large numbers of leukocytes, the majority of which are of myeloid lineage, and mostly neutrophiles in acute settings, whereas macrophages and dendritic cells predominate in chronic settings. In the case of chronic immune-mediated diseases, T lymphocytes are predominant
[66].
Activated macrophages may either damage the tissue directly or generate profibrotic cytokines, including TGF-β and other growth factors, and are capable of producing some matrix constituents. It is therefore evident that fibrosis and renal inflammation, primarily driven by immune system activation, are closely related.
Beyond its role in immune function regulation, AA is also directly related to fibrosis. In vitro experiments of cell cultures incubated with PUFAs showed that AA is able to induce upregulation of the expression of TGF-β, fibronectin 1 (FN1), connective tissue growth factor (CTGF), and collagen IV, all compounds related to fibrosis
[67]. AA also enhances in vitro angiotensin II (AngII)-induced gene expression
[67], activating mechanisms that mediate renal damage. Interestingly, omega-3 EPA and DHA, if administered with AA, suppress the effects of both AA and AngII
[67]. On the other side, angiotensin II is degraded to form angiotensin-(1-7), which inhibits angiotensin II-stimulated phosphorylation of the mitogen-activated protein kinases (MAPKs) p38, extracellular signal-related kinase (ERK1/ERK2), and C-JUN N-terminal kinase (JNK) in proximal tubular cells, thus exerting a protective role against fibrosis. As a matter of fact, the p38 MAPK phosphorylation leads to the release of AA and the production of TGF-β 1 and extracellular matrix proteins
[68].
20-HETE, an AA metabolite, also plays a distinct role in fibrogenesis, by activating the renin-angiotensin-aldosterone system (RAAS), by inducing vascular expression of ACE downstream of NF-κB activation
[69][70]. It is well known that the RAAS is involved in renal fibrosis
[71], because it increases TGF-β expression, which starts a biomolecular cascade driving to renal fibrosis.
On the contrary, PGE
2, another AA metabolite, has been shown to inhibit collagen type 1 production and to induce matrix metalloproteinase 1 (MMP1) expression in dermal fibroblasts
[72] by binding to the EP-1 receptor on fibroblasts, starting a pathway-regulated ERK1/2 and IP3 signaling that leads to a reduction in collagen expression and an increase in MMP1 expression
[72].
8. Drug and Gene Interactions
Idiopathic nephrotic syndrome is usually treated with glucocorticoids or with immunosuppressive drugs, particularly calcineurine inhibitors (CNI), such as cyclosporine A (CsA) and tacrolimus (Fk). CNIs are metabolized mainly by cytochrome P450, encoded by the CYP gene cluster. As seen above, the CYP gene is also involved in AA metabolism, but in the literature, there are no reports of enzymatic competition between these drugs and AA.
With regard to the relationship between CNI and AA blood levels, an in vitro study reported that CsA decreases the activity of Delta 9 desaturase and increases the activity of Delta 6 and Delta 5 desaturases
[73] through unknown mechanisms. However, as Delta 5 desaturase is involved in the last step of AA biosynthesis
[73], CsA therapy could increase AA blood level of patients with INS.
On the same line, it has been suggested that CsA mostly increased the availability of free AA instead of decreasing AA blood levels through the acceleration of AA conversion by the cyclooxygenase pathway
[74], but a further in vitro study concluded that CsA had no effect on AA release and metabolism
[75]. This result was confirmed more recently in a study of CsA and glucocorticosteroids in human peripheral blood mononuclear cells
[76].
With regard to the role of AA metabolism in determining CNI side effects, it is well known that CsA treatment may cause gingival overgrowth, which depends on PGE
2 production in gingival fibroblasts. In fact, CsA potentiates TNF-α to stimulate the release of AA from fibroblasts, with consequent enhanced production of PGE
2 and gingival overgrowth
[77]. There are no studies reporting the same effect in other tissues.
The nephrotoxicity of CsA is well established, and Fk administration is associated with the same side effect, which has been linked to CYP2C8*3 and CYP2C8*4 polymorphisms and a consequent reduction of EETs: it was observed that a circulating Fk plasma concentration of 10 ng/mL is able to reduce the production of eicosanoids by 35%. It follows that CNIs-induced nephrotoxicity could be due to a reduced activity of CYP2C8*3, which reduces the production of EETs, enhancing drug nephrotoxicity
[78].
Pre-treatment with Fk is also known to enhance glucocorticoids to inhibit AA and PGE
2 production
[79] by inhibiting COX2 expression, but the co-administration of Fk and glucocorticoids does not inhibit COX2 expression, allowing for normal PGE
2 production
[79].
9. Dietary Balance Between AA and LA and AA Sources
AA, which belongs to the omega-6 series, and docosahexaenoic acid (DHA), which belongs to the omega-3 series, are the most important byproducts of essential fatty acids linoleic and α-linolenic acid, and their imbalance has been associated with inflammatory and chronic disorders
[80].
While LA and AA are mostly known as inflammatory molecules, operating within an interdependent network through their metabolites
[80], AA metabolites also have anti-inflammatory and protective roles, while LA metabolites affect immune function by binding cellular receptors and altering signaling molecules
[81].
AA and DHA levels depend on both to genetic predisposition and diet intake. Blood AA levels, as shown in , can be modulated through dietary habits, taking into account that there is a marked difference between the amount of AA supplied with the diet and the amount synthesized by human metabolic pathways. In the latter case, the main rate-limiting enzymes are the Δ5- and Δ6-desaturases, which are encoded by the genes FADS1 and FADS2, and different polymorphisms in the fatty acid desaturases genes might even increase or decrease the production of these LC-PUFAs
[82]. As a matter of fact, looking at the frequencies of the 28 SNPs in the FADS haplotypes, their distribution in the 3 main haplotypes is evident over the world
[83].
Unlike other fatty acids, omega-3 and omega-6 precursors (LA and linolenic acid, respectively) cannot be synthesized de novo by mammals (they are essential dietary compounds indeed), so the relative abundance of these PUFAs in the diet has a major influence in humans.
LA is the most represented omega-6 PUFA in most western diets, and is widely distributed in foods: it represents more than 50% of the lipid content in various vegetable oils, including safflower, sunflower, corn, and soybean oils; it is present in high amounts in nuts and seeds, while lower levels are found in whole grains, legumes, some meats, eggs, and dairy products
[84]. Notably, it was recently reported that a strong reduction in dietary intake of LA was not associated with a linear decrease in circulating AA levels
[85].
The AA state depends on the endogenous synthesis from the essential precursor LA, undergoing desaturation and elongation, and the direct dietary intake
[86]. Since LA to AA conversion efficiency is low in humans, AA intake through the diet appears to be significantly more effective in raising its circulating levels.
 +1 credit
+1 credit