Studies concerning the role of arachidonic acid (AA) and its metabolites in kidney disease are scarce, and this applies in particular to idiopathic nephrotic syndrome (INS). INS is one of the most frequent glomerular diseases in childhood; it is characterized by T-lymphocyte dysfunction, alterations of pro- and anti-coagulant factor levels, and increased platelet count and aggregation, leading to thrombophilia. AA and its metabolites are involved in several biological processes. Herein, we describe the main fields where they may play a significant role, particularly as it pertains to their effects on the kidney and the mechanisms underlying INS. AA and its metabolites influence cell membrane fluidity and permeability, modulate platelet activity and coagulation, regulate lymphocyte activity and inflammation, preserve the permeability of the glomerular barrier, influence podocyte physiology, and play a role in renal fibrosis.
- kidney
- arachidonic acid
- nephrotic syndrome
1. Introduction
| Neonates | Children | Adults | Elderly | |
|---|---|---|---|---|
| Linoleic acid (%) | 4.61 ± 1.06 | 17.67 ± 1.92 | 18.41 ± 2.87 | 17.64 ± 2.89 |
| Arachidonic acid (%) | 13.14 ± 1.73 | 8.33 ± 1.04 | 8.51 ± 1.38 | 8.32 ± 1.40 |
| Total omega 6 (%) | 22.99 ± 2.13 | 28.97 ± 2.19 | 29.79 ± 3.13 | 28.78 ± 3.24 |
| Total saturated fatty acids (%) | 46.10 ± 3.16 | 44.32 ± 1.61 | 39.47 ± 2.3 | 39.83 ± 2.16 |
| Total monounsaturated fatty acids (%) | 26.15 ± 2.76 | 24.39 ± 2.07 | 27.20 ± 3.08 | 27.83 ± 3.27 |
4. Immune System
The therapeutic efficacy of Rituximab in modifying the course of steroid-dependent nephrotic syndrome suggested that B cells play a key role in the pathogenesis of INS. This was recently confirmed by evidence of a pathological increase in memory B cells in INS [25]. Moreover, other studies showed a decrease of Treg cells [26], dysregulation of T-cells [27], lower levels of NK and NKT cells, and increased levels of inflammatory markers during proteinuria [28,29][28][29]. These studies confirm [1] that the immune system plays a pivotal role in non-genetic INS and specifically in the loss of the glomerular barrier function, by activating the inflammatory process against podocytes. In immune cells, like lymphocytes, neutrophils, and monocytes, AA constitutes about 20% of total fatty acids, while EPA and DHA constitute 1% and 2.5%, respectively [30]. It was reported that oral administration of omega-3 fatty acids changes the pattern of production of eicosanoids, by increasing resolvins production, thus affecting phagocytosis, T-cell signaling, and antigen presentation capability. These effects seem to be mediated at the membrane level [30]. The distribution of AA within intracellular lipid pools in inflammatory cells has an important role in regulating eicosanoids production. In fact, a pool of AA was identified within the triglycerides of mast cells, eosinophils, monocytes, and platelets [31]. When inflammatory cells are activated, AA is released from membrane phospholipids into the cell and partially incorporated into intracellular triglycerides, ready to supply membrane phospholipids again after cell activation has ended [32]. Thus, AA metabolites can act in several ways on lymphocyte activity, affecting inflammation levels [32,33,34][32][33][34] (Table 2) and possibly the course of INS.| Cell Type | AA Metabolite | Effect | |
|---|---|---|---|
| Basophil | PGD | 2 | Stimulates basophil chemotaxis |
| Eosinophil | PGD | 2 | |
| Total omega 3 (%) | |||
| 4.76 ± 0.89 | 2.31 ± 0.50 | 3.54 ± 1.05 | 3.55 ± 0.95 |
2. Cell Membrane Fluidity and Permeability
It was recently described that erythrocyte membranes of patients with INS differ from those of normal subjects, particularly due to reduced membrane fluidity [11]. AA is one of the most abundant fatty acids in the cell membrane, to which it endows mobility and flexibility [12,13][12][13]. The fatty acid composition determines the viscosity of the cell lipid bilayer and membrane fluidity, thus directly affecting the function of specific membrane proteins, like, for example, those involved in cellular inflammatory signaling, namely lymphocyte function-associated antigen 1 (LFA-1), intercellular adhesion molecule 1 (ICAM-1), and cluster of differentiation 2 (CD2) [12,13][12][13]. With regard to membrane permeability, AA acts on Ca2+ cell load [10] with a double effect: at low micromolar concentrations it increases Ca2+-ATPase activity, while at higher concentrations it reduces ATPase activity. This may be due to an unspecific and non-physiological inhibitory effect on the hydrolytic activity of P-type ATPase. ATPases are a superfamily of lipid pumps involved, among other functions, in secretion and absorption at the kidney level; these pumps are blocked by protein kinase C inhibitors [14]. AA increases membrane permeability to calcium, which is a key factor for platelet activation [15]. AA may act on ion channels by either binding to or inserting among the membrane molecules, thus modifying the mechanical properties of the cell membrane and modulating channel function [16]. AA also has a direct effect on several membrane potassium channels, either by accelerating their inactivation (in particular, the A-type channels and delayed rectifier channels), or by inducing the activation of large-conductance voltage-independent channels. The two-pore domain potassium channels are inactivated by AA as well, in contrast to what usually occurs with classical K channel-blocking drugs [16]. Transient receptor potential channels (TPR) are instead activated directly by AA and its lipoxygenase (LOX)-derived metabolites [16] (namely, 12- and 15-(S)-hydroperoxyeicosatetraenoic acids, 5- and 15-(S)-hydroxyeicosatetraenoic acids, and leukotriene B4). LOX metabolites can activate the TPR channel by virtue of their structure that mimics the capsaicin structure [17]. Interestingly, AA and its metabolic byproducts effects on calcium and potassium balance at the membrane level have been hypothesized to underlie the molecular-related derangements in INS [6]. As it concerns membrane fluidity, albumin is the main fatty acid-binding protein in extracellular fluid, having seven fatty acid-binding sites [18]. Albumin increases AA release from cell membranes in a concentration-dependent manner, by interacting with membrane phospholipids on the extracellular surface; in particular, positively charged arginine residues at or near albumin’s binding sites for LCFA interact with AA, determining its release from the phospholipid layer [19]. Thus, albumin decreases cell membrane permeability of endothelial and circulating cells to water and small solutes [19].3. Platelet Aggregation and Coagulation
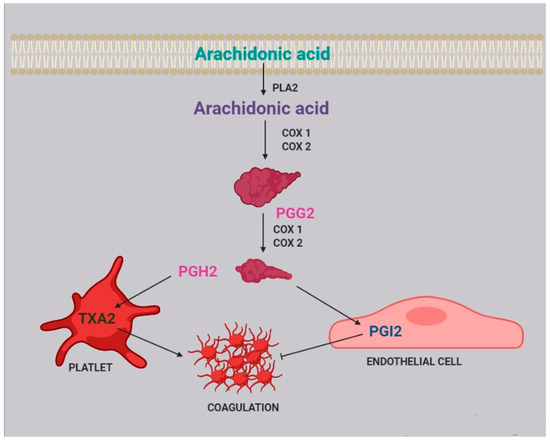
Two of the most active compounds related to platelet function are thromboxane and prostacyclin, both metabolites of AA [20]. AA is released from platelet membrane by phospholipase A2 (PLA2), which hydrolyzes the bond between the second fatty acid of phospholipids and the glycerol molecule. The released AA is then metabolized by cyclooxygenase [21], generating prostaglandin G2, and thereafter prostaglandin H2. Afterwards, two different pathways can take place: the first one, within the platelets, leads to the synthesis of thromboxane A2 (TXA2) and subsequently B2 (TXB2); the second one, within the endothelial cells, leads to the synthesis of prostacyclin (PGI2) (Figure 2).| Stimulates eosinophil chemotaxis | |||
| Blocks eosinophil apoptosis | |||
| Activates eosinophils | |||
| Naive t cell | TXA | 2 | Inhibits proliferation of naive T cells |
| B-cell | PGE | 2 | Enhances IgE class switching by B cells |
| Dendritic cell | LTB | 4 | Stimulates DC production of IL-6 |
| LTC | 4 | Participates in cell migration | |
| Enhances cells activation and functions | |||
| PGD | 2 | Inhibits cells migration | |
| PGE | 2 | Stimulates IL-10 production | |
| Modulates cell migration | |||
| Downregulates major histocompatibility complex C class II expression | |||
| Inhibits IL-12 and IFN-ã production | |||
| Inhibits the expression of CCL3/CCL4 | |||
| PGJ | 2 | Induces apoptosis | |
| TXA | 2 | Inhibits interaction with T cell | |
| Langerhans cell | PGE | 2 | Promotes the migration and maturation of Langerhans cells |
| PGD | 2 | Inhibits cells migration | |
| Lymphocyte | PGE | 2 | Inhibits interactions with endothelial cell |
| Macrophage | PGE | 2 | Suppresses cytokine production |
| Suppresses chemokine expression | |||
| PGJ | 2 | Inhibits release of IL-10 and IL-12 | |
| Mast cell | PGE | 2 | Enhances antigen-stimulated degranulation |
| Neutrophil | LTB | 4 | Activates cells |
| NK cell | PGE | 2 | Inhibits IL-12 and IFN-ã production |
| T cell | LTB | 4 | Enhances cell recruitment |
| PGE | 2 | Inhibits cell proliferation | |
| TXA2 | Inhibits interactions with dendritic cells | ||
| Regulates the elimination of self-reactive cells | |||
| Increases cell proliferation and activation | |||
| Enhances local cytotoxic cell function | |||
| Th2 | PGD | 2 | Stimulates chemotaxis |
5. Kidney Glomerular and Tubular Function
6. Podocyte Physiopathology and Infections
It is well known that, in the course of nephrotic syndrome, infections lead to an exacerbation of proteinuria [55] and are an important risk factor for relapses [1,56,57][1][56][57]. Infections activate the immune system, triggering the inflammatory cascade. It was recently reported that during inflammation two enzymes are induced: 15-lipoxygenase (15-LO) and secreted phospholipase A2 (sPLA2) [58]. Notably, 15-LO is expressed in human podocytes [59], while sPLA2 is expressed in platelets, neutrophils, eosinophils, and macrophages [60]. sPLA2 releases AA from membrane phospholipids [51,61][51][61] acting in a paracrine way. In glomerular podocytes, intracellular free AA is metabolized to PGE2, which, by interacting with the EP 4 receptor (prostaglandin E2 receptor 4) expressed by podocytes, reduces AA release [52]. This loop regulates podocyte function in both physiological and pathological conditions and is able to change PGE2 synthesis [62]. As described above, during the course of an infection, intracellular AA levels increase due to the action of sPLA2. It was reported that in podocytes, an excess of AA activates protein kinase A, which in turn promotes c-Abl activation and nephrin phosphorylation, thus causing actin cytoskeleton remodeling and podocyte injury [63]. This mechanism could partially explain the frequent recurrence of proteinuria during infectious episodes in children. Moreover, an increase in sPLA2 1B levels and PLA2R expression has been observed to be positively associated to podocyte apoptosis in kidneys of patients with idiopathic membranous nephropathy [64]. Podocyte foot process injury and podocyte apoptosis due to cytoskeleton remodeling were also attributed to a change of Ca2+ efflux [51], driven by 20-HETE, the main AA metabolite. It has also been observed that 20-HETE increases the current Ca2+ flowing through TRPC6 channels in the podocyte [51], which are located at the slit diaphragm, possibly leading to cellular injury.7. Renal Fibrosis
Renal fibrosis is a process that progresses independently of the primary renal disease [65] and represents a failed wound-healing process of the kidney tissue. Renal biopsies of patients with steroid-resistant nephrotic syndrome often show glomerulosclerosis and interstitial fibrosis, which are associated with progression to end-stage kidney disease in more than 50% of cases [65], a poor prognosis that heightens the necessity of increasing our knowledge of the mechanisms underlying fibrosis. Renal fibrosis is characterized by connective tissue deposition in the kidney parenchyma, particularly in the interstitial space and within the walls of glomerular capillaries, and by the consequent cellular processes. Fibrosis also interferes with normal tubular function, leading progressively to organ failure [65,66][65][66]. The scar tissue contains fibrillar collagen I and III as well as some constituents of the normal capillary basement membrane, like collagen IV and V, fibronectin, laminin, perlecan, and heparin [66]. Fibrosis is associated with leukocyte recruitment, angiogenesis, vascular leak, and the appearance of myofibroblasts. In particular, both the glomerulus and the interstitium attract large numbers of leukocytes, the majority of which are of myeloid lineage, and mostly neutrophiles in acute settings, whereas macrophages and dendritic cells predominate in chronic settings. In the case of chronic immune-mediated diseases, T lymphocytes are predominant [66]. Activated macrophages may either damage the tissue directly or generate profibrotic cytokines, including TGF-β and other growth factors, and are capable of producing some matrix constituents. It is therefore evident that fibrosis and renal inflammation, primarily driven by immune system activation, are closely related. Beyond its role in immune function regulation, AA is also directly related to fibrosis. In vitro experiments of cell cultures incubated with PUFAs showed that AA is able to induce upregulation of the expression of TGF-β, fibronectin 1 (FN1), connective tissue growth factor (CTGF), and collagen IV, all compounds related to fibrosis [67]. AA also enhances in vitro angiotensin II (AngII)-induced gene expression [67], activating mechanisms that mediate renal damage. Interestingly, omega-3 EPA and DHA, if administered with AA, suppress the effects of both AA and AngII [67]. On the other side, angiotensin II is degraded to form angiotensin-(1-7), which inhibits angiotensin II-stimulated phosphorylation of the mitogen-activated protein kinases (MAPKs) p38, extracellular signal-related kinase (ERK1/ERK2), and C-JUN N-terminal kinase (JNK) in proximal tubular cells, thus exerting a protective role against fibrosis. As a matter of fact, the p38 MAPK phosphorylation leads to the release of AA and the production of TGF-β 1 and extracellular matrix proteins [68]. 20-HETE, an AA metabolite, also plays a distinct role in fibrogenesis, by activating the renin-angiotensin-aldosterone system (RAAS), by inducing vascular expression of ACE downstream of NF-κB activation [69,70][69][70]. It is well known that the RAAS is involved in renal fibrosis [71], because it increases TGF-β expression, which starts a biomolecular cascade driving to renal fibrosis. On the contrary, PGE2, another AA metabolite, has been shown to inhibit collagen type 1 production and to induce matrix metalloproteinase 1 (MMP1) expression in dermal fibroblasts [72] by binding to the EP-1 receptor on fibroblasts, starting a pathway-regulated ERK1/2 and IP3 signaling that leads to a reduction in collagen expression and an increase in MMP1 expression [72].8. Drug and Gene Interactions
Idiopathic nephrotic syndrome is usually treated with glucocorticoids or with immunosuppressive drugs, particularly calcineurine inhibitors (CNI), such as cyclosporine A (CsA) and tacrolimus (Fk). CNIs are metabolized mainly by cytochrome P450, encoded by the CYP gene cluster. As seen above, the CYP gene is also involved in AA metabolism, but in the literature, there are no reports of enzymatic competition between these drugs and AA. With regard to the relationship between CNI and AA blood levels, an in vitro study reported that CsA decreases the activity of Delta 9 desaturase and increases the activity of Delta 6 and Delta 5 desaturases [73] through unknown mechanisms. However, as Delta 5 desaturase is involved in the last step of AA biosynthesis [73], CsA therapy could increase AA blood level of patients with INS. On the same line, it has been suggested that CsA mostly increased the availability of free AA instead of decreasing AA blood levels through the acceleration of AA conversion by the cyclooxygenase pathway [74], but a further in vitro study concluded that CsA had no effect on AA release and metabolism [75]. This result was confirmed more recently in a study of CsA and glucocorticosteroids in human peripheral blood mononuclear cells [76]. With regard to the role of AA metabolism in determining CNI side effects, it is well known that CsA treatment may cause gingival overgrowth, which depends on PGE2 production in gingival fibroblasts. In fact, CsA potentiates TNF-α to stimulate the release of AA from fibroblasts, with consequent enhanced production of PGE2 and gingival overgrowth [77]. There are no studies reporting the same effect in other tissues. The nephrotoxicity of CsA is well established, and Fk administration is associated with the same side effect, which has been linked to CYP2C8*3 and CYP2C8*4 polymorphisms and a consequent reduction of EETs: it was observed that a circulating Fk plasma concentration of 10 ng/mL is able to reduce the production of eicosanoids by 35%. It follows that CNIs-induced nephrotoxicity could be due to a reduced activity of CYP2C8*3, which reduces the production of EETs, enhancing drug nephrotoxicity [78]. Pre-treatment with Fk is also known to enhance glucocorticoids to inhibit AA and PGE2 production [79] by inhibiting COX2 expression, but the co-administration of Fk and glucocorticoids does not inhibit COX2 expression, allowing for normal PGE2 production [79].9. Dietary Balance Between AA and LA and AA Sources
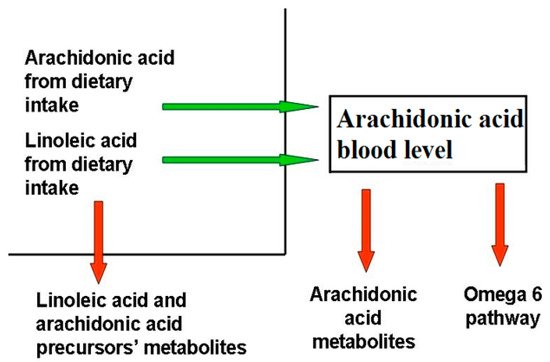
AA, which belongs to the omega-6 series, and docosahexaenoic acid (DHA), which belongs to the omega-3 series, are the most important byproducts of essential fatty acids linoleic and α-linolenic acid, and their imbalance has been associated with inflammatory and chronic disorders [82][80]. While LA and AA are mostly known as inflammatory molecules, operating within an interdependent network through their metabolites [82][80], AA metabolites also have anti-inflammatory and protective roles, while LA metabolites affect immune function by binding cellular receptors and altering signaling molecules [83][81]. AA and DHA levels depend on both to genetic predisposition and diet intake. Blood AA levels, as shown in Figure 1, can be modulated through dietary habits, taking into account that there is a marked difference between the amount of AA supplied with the diet and the amount synthesized by human metabolic pathways. In the latter case, the main rate-limiting enzymes are the Δ5- and Δ6-desaturases, which are encoded by the genes FADS1 and FADS2, and different polymorphisms in the fatty acid desaturases genes might even increase or decrease the production of these LC-PUFAs [84][82]. As a matter of fact, looking at the frequencies of the 28 SNPs in the FADS haplotypes, their distribution in the 3 main haplotypes is evident over the world [85][83]. Unlike other fatty acids, omega-3 and omega-6 precursors (LA and linolenic acid, respectively) cannot be synthesized de novo by mammals (they are essential dietary compounds indeed), so the relative abundance of these PUFAs in the diet has a major influence in humans. LA is the most represented omega-6 PUFA in most western diets, and is widely distributed in foods: it represents more than 50% of the lipid content in various vegetable oils, including safflower, sunflower, corn, and soybean oils; it is present in high amounts in nuts and seeds, while lower levels are found in whole grains, legumes, some meats, eggs, and dairy products [86][84]. Notably, it was recently reported that a strong reduction in dietary intake of LA was not associated with a linear decrease in circulating AA levels [87][85]. The AA state depends on the endogenous synthesis from the essential precursor LA, undergoing desaturation and elongation, and the direct dietary intake [88][86]. Since LA to AA conversion efficiency is low in humans, AA intake through the diet appears to be significantly more effective in raising its circulating levels.References
- Noone, D.G.; Iijima, K.; Parekh, R. Idiopathic Nephrotic Syndrome in Children. Lancet 2018, 392, 61–74.
- Teoh, C.W.; Robinson, L.A.; Noone, D. Perspectives on edema in childhood nephrotic syndrome. Am. J. Physiol. Ren. Physiol. 2015, 309, F575–F582.
- Dorhout Mees, E.J.; Koomans, H.A. Understanding the nephrotic syndrome: What’s new in a decade? Nephron 1995, 70, 1–10.
- Eneman, B.; Levtchenko, E.; van den Heuvel, B.; Van Geet, C.; Freson, K. Platelet abnormalities in nephrotic syndrome. Pediatr. Nephrol. 2016, 31, 1267–1279.
- Vivarelli, M.; Massella, L.; Ruggiero, B.; Emma, F. Minimal Change Disease. Clin. J. Am. Soc. Nephrol. 2017, 12, 332–345.
- Ranganathan, S. Pathology of Podocytopathies Causing Nephrotic Syndrome in Children. Front. Pediatr. 2016, 4, 32.
- Eddy, A.A.; Symons, J.M. Nephrotic Syndrome in Childhood. Lancet 2003, 362, 629–639.
- Risé, P.; Tragni, E.; Ghezzi, S.; Agostoni, C.; Marangoni, F.; Poli, A.; Catapano, A.L.; Siani, A.; Iacoviello, L.; Galli, C. Different patterns characterize Omega 6 and Omega 3 long chain polyunsaturated fatty acid levels in blood from Italian infants, children, adults and elderly. Prostagland. Leukot. Essent. Fat. Acids 2013, 89, 215–220.
- Cunnnane, S.C.; Kent, E.T.; McAdoo, K.R.; Caldwell, D.; Lin, A.N.; Carter, D.M. Abnormalities of Plasma and Erythrocyte Essential Fatty Acid Composition in Epidermolysis Bullosa: Influence of Treatment with Diphenylhydantoin. J. Investig. Dermatol. 1987, 89, 395–399.
- Strandvik, B. Fatty acid metabolism in cystic fibrosis. Prostagland. Leukot. Essent. Fat. Acids 2010, 83, 121–129.
- Cheng, S.; Chen, Y.L. Study on erythrocyte membrane fluidity by laser Raman spectroscopy. Cell Biol. Int. Rep. 1988, 12, 205–211.
- Pompéia, C.; Lopes, L.R.; Miyasaka, C.K.; Procópio, J.; Sannomiya, P.; Curi, R. Effect of fatty acids on leukocyte function. Braz. J. Med. Biol. Res. 2000, 33, 1255–1268.
- Brash, A.R. Arachidonic acid as a bioactive molecule. J. Clin. Investig. 2001, 107, 1339–1345.
- Oliveira, V.H.; Nascimento, K.S.; Freire, M.M.; Moreira, O.C.; Scofano, H.M.; Barrabin, H.; Mignaco, J.A. Mechanism of modulation of the plasma membrane Ca(2+)-ATPase by arachidonic acid. Prostagland. Other Lipid Mediat. 2008, 87, 47–53.
- Varga-Szabo, D.; Braun, A.; Nieswandt, B. Calcium signaling in platelets. J. Thromb. Haemost. 2009, 7, 1057–1066.
- Meves, H. Arachidonic acid and ion channels: An update. Br. J. Pharmacol. 2008, 155, 4–16.
- Hwang, S.W.; Cho, H.; Kwak, J.; Lee, S.Y.; Kang, C.J.; Jung, J.; Cho, S.; Min, K.H.; Suh, Y.G.; Kim, D.; et al. Direct activation of capsaicin receptors by products of lipoxygenases: Endogenous capsaicin-like substances. Proc. Natl. Acad. Sci. USA 2000, 97, 6155–6160.
- Van der Vusse, G.J. Albumin as fatty acid transporter. Drug Metab. Pharmacokinet. 2009, 24, 300–307.
- Beck, R.; Bertolino, S.; Abbot, S.E.; Aaronson, P.I.; Smirnov, S.V. Modulation of arachidonic acid release and membrane fluidity by albumin in vascular smooth muscle and endothelial cells. Circ. Res. 1998, 83, 923–931.
- Nelson, G.J.; Schmidt, P.C.; Bartolini, G.; Kelley, D.S.; Kyle, D. The effect of dietary arachidonic acid on platelet function, platelet fatty acid composition, and blood coagulation in humans. Lipids 1997, 32, 421–425.
- Trostchansky, A.; Moore-Carrasco, R.; Fuentes, E. Oxydative pathways of arachidonic acid as targets for regulation of platelet activation. Prostaglandines Other Lipid Mediat. 2019, 15, 106382.
- Offermanns, S. Activation of platelet function through G protein-coupled receptors. Circ. Res. 2006, 99, 1293–1304.
- Rasedee, A.; Feldman, B.F. Nephrotic syndrome: A platelet hyperaggregability state. Vet. Res. Commun. 1985, 9, 199–211.
- Davì, G.; Averna, M.; Catalano, I.; Barbagallo, C.; Ganci, A.; Notarbartolo, A.; Ciabattoni, G.; Patrono, C. Increased thromboxane biosynthesis in type IIa hypercholesterolemia. Circulation 1992, 85, 1792–1798.
- Colucci, M.; Carsetti, R.; Cascioli, S.; Serafinelli, J.; Emma, F.; Vivarelli, M. B cell phenotype in pediatric idiopathic nephrotic syndrome. Pediatr. Nephrol. 2019, 34, 177–181.
- Chebotareva, N.; Bobkova, I.; Lysenko, L. T regulatory cells in renal tissue of patients with nephrotic syndrome. Pediatr. Int. 2020, 62, 884–885.
- Chambers, E.T.; Gbadegesin, R.A. Aberrant IgM on T cells: Biomarker or pathogenic factor in childhood nephrotic syndrome? Kidney Int. 2019, 96, 818–820.
- Guimarães, F.T.L.; Melo, G.E.B.A.; Cordeiro, T.M.; Feracin, V.; Vieira, E.R.; Pereira, W.F.; Pinheiro, S.V.B.; Miranda, A.S.; Simões-E-Silva, A.C. T-lymphocyte-expressing inflammatory cytokines underlie persistence of proteinuria in children with idiopathic nephrotic syndrome. J. Pediatr. 2018, 94, 546–553.
- Alsharidah, A.S.; Alzogaibi, M.A.; Bayoumy, N.M.; Alghonaim, M. Neutrophil chemokines levels in different stages of nephrotic syndrome. Saudi J. Kidney Dis. Transplant. 2017, 28, 1256–1263.
- Calder, P.C. The relationship between the fatty acid composition of immune cells and their function. Prostagland. Leukot. Essent. Fat. Acids 2008, 79, 101–108.
- Triggiani, M.; Oriente, A.; de Crescenzo, G.; Rossi, G.; Marone, G. Biochemical functions of a pool of arachidonic acid associated with triglycerides in human inflammatory cells. Int. Arch. Allergy Immunol. 1995, 107, 261–263.
- Wei, J.; Gronert, K. Eicosanoid and specialized preresolving mediator regulation of lymphoid cells. Trends Biochem. Sci. 2019, 44, 214–225.
- Hanna, V.S.; Hafez, E.A. Synopsis of arachidonic acid metabolism—A review. J. Adv. Res. 2018, 11, 23–32.
- Wang, T.; Fu, X.; Chen, Q.; Part, J.K.; Wang, D.; Wang, Z.; Gai, Z. Arachidonic acid metabolism and kidney inflammation. Int. J. Mol. Sci. 2019, 20, 3683.
- Nataraj, C.; Thomas, D.W.; Tilley, S.L.; Nguyen, M.T.; Mannon, R.; Koller, B.H.; Coffman, T.M. Receptors for prostaglandin E(2) that regulate cellular immune responses in the mouse. J. Clin. Investig. 2001, 108, 1229–1235.
- Smith, W.L. The eicosanoids and their biochemical mechanisms of action. Biochem. J. 1989, 259, 315–324.
- Legler, D.F.; Bruckner, M.; Uetz-von Allmen, E.; Krause, P. Prostaglandin E2 at new glance: Novel insights in functional diversity offer therapeutic chances. Int. J. Biochem. Cell Biol. 2010, 42, 198–201.
- Harizi, H.; Gualde, N. The impact of eicosanoids on the crosstalk between innate and adaptive immunity: The key roles of dendritic cells. Tissue Antigens 2005, 65, 507–514.
- Roper, R.L.; Phipps, R.P. Prostaglandin E2 and cAMP inhibit B lymphocyte activation and simultaneously promote IgE and IgG1 synthesis. J. Immunol. 1992, 149, 2984–2991.
- Goodarzi, K.; Goodarzi, M.; Tager, A.M.; Luster, A.D.; von Andrian, U.H. Leukotriene B4 and BLT1 control cytotoxic effector T cell recruitment to inflamed tissues. Nat. Immunol. 2003, 4, 965–973.
- Yang, L.; Mäki-Petäjä, K.; Cheriyan, J.; McEniery, C.; Wilkinson, I.B. The role of epoxyeicosatrienoic acids in the cardiovascular system. Br. J. Clin. Pharmacol. 2015, 80, 28–44.
- Fang, Q.; Chen, G.Z.; Wang, Y.; Wang, D.W. Role of cytochrome P450 epoxygenase-dependent arachidonic acid metabolites in kidney physiology and diseases. Sheng Li Xue Bao 2018, 70, 591–599.
- Wang, D.; DuBois, R.N. Epoxyeicosatrienoic acids: A double-edged sword in cardiovascular diseases and cancer. J. Clin. Investig. 2012, 122, 19–22.
- Fleming, I. Vascular cytochrome p450 enzymes: Physiology and pathophysiology. Trends Cardiovasc. Med. 2008, 18, 20–25.
- Kim, J.; Yoon, S.P.; Toews, M.L.; Imig, J.D.; Hwang, S.H.; Hammock, B.D.; Padanilam, B.J. Pharmacological inhibition of soluble epoxide hydrolase prevents renal interstitial fibrogenesis in obstructive nephropathy. Am. J. Physiol. Ren. Physiol. 2015, 308, F131–F139.
- Imig, J.D.; Hammock, B.D. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat. Rev. Drug Discov. 2009, 8, 794–805.
- Carroll, M.A.; Cheng, M.K.; Liclican, E.L.; Li, J.; Doumad, A.B.; McGiff, J.C. Purinoceptors in renal microvessels: Adenosine-activated and cytochrome P450 monooxygenase-derived arachidonate metabolites. Pharmacol. Rep. 2005, 57, 191–195.
- Williams, J.M.; Sharma, M.; Anjaiahh, S.; Falck, J.R.; Roman, R.J. Role of endogenous CYP450 metabolites of arachidonic acid in maintaining the glomerular protein permeability barrier. Am. J. Physiol. Ren. Physiol. 2007, 293, F501–F505.
- Fan, F.; Roman, R.J. Effect of Cytochrome P450 Metabolites of Arachidonic Acid in Nephrology. J. Am. Soc. Nephrol. 2017, 28, 2845–2855.
- Nowicki, S.; Chen, S.L.; Aizman, O.; Cheng, X.J.; Li, D.; Nowicki, C.; Nairn, A.; Greengard, P.; Aperia, A. 20-Hydroxyeicosa-tetraenoic acid (20 HETE) activates protein kinase C. Role in regulation of rat renal Na+,K+-ATPase. J. Clin. Investig. 1997, 99, 1224–1230.
- Roshanravan, H.; Kim, E.Y.; Dryer, S.E. 20-Hydroxyeicosatetraenoic Acid (20-HETE) Modulates in Podocytes. Front. Physiol. 2016, 7, 351.
- Shatat, I.F.; Becton, L.J.; Woroniecki, R.P. Hypertension in childhood nephrotic syndrome. Front. Pediatr. 2019, 7, 287.
- Quigley, R.; Baum, M.; Reddy, K.M.; Griener, J.C.; Falck, J.R. Effects of 20-HETE and 19(s)-HETE on rabbit proximal straight tubule volume transport. Am. J. Physiol. Ren. Physiol. 2000, 278, 949–953.
- Osama, H.E.; Sheriff, M.S.; Anwar, M.; Ayman, O.S. Clinical implication of 20-hydroeicosatetraenoic acid in the kidney, liver, lung and brain. An emerging therapeutic target. Pharmaceutics 2017, 9, 9.
- Han, H.; Wang, S.; Liang, Y.; Lin, J.; Shi, L.; Ye, L.; Song, S.; He, M.; Li, S.; Chen, F.; et al. Respiratory tract infection: A risk factor for the onset and relapse of adult onset minimal changes disease in Southern China. BioMed Res. Int. 2018, 2018, 1657208.
- Mishra, O.P.; Abhinay, A.; Mishra, R.N.; Prasad, R.; Pohl, M. Can we predict relapses in children with idiopathic steroid-sensitive nephrotic syndrome? J. Trop. Pediatr. 2013, 59, 343–349.
- Manta, M.; Singh, S. Infection associated relapses in children with nephrotic syndrome: A short-term outcome study. Saudi J. Kidney Dis. Transplant. 2019, 30, 1245–1253.
- Ha, V.T.; Lainšček, D.; Gesslbauer, B.; Jarc-Jovičić, E.; Hyötyläinen, T.; Ilc, N.; Lakota, K.; Tomšič, M.; van de Loo, F.A.J.; Bochkov, V.; et al. Synergy between 15-lipoxygenase and secreted PLA2 promotes inflammation by formation of TLR4 agonists from extracellular vesicles. Proc. Natl. Acad. Sci. USA 2020, 117, 25679–25689.
- Yiu, S.S.; Zhao, X.; Inscho, E.W.; Imig, J.D. 12-Hydroxyeicosatetraenoic acid participates in angiotensin II afferent arteriolar vasoconstriction by activating L-type calcium channels. J. Lipid Res. 2003, 44, 2391–2399.
- Andreani, M.; Olivier, J.L.; Berenbaum, F.; Raymondjean, M.; Béréziat, G. Transcriptional regulation of inflammatory secreted phospholipases A(2). Biochim. Biophys. Acta 2000, 1488, 149–158.
- Ren, X.; Zhang, M.; Chen, L.; Zhang, W.; Huang, Y.; Luo, H.; Li, L.; He, H. The anti-inflammatory effects of Yunnan Baiyao are involved in regulation of the phospholipase A2/arachidonic acid metabolites pathways in acute inflammation rat model. Mol. Med. Rep. 2017, 16, 4045–4053.
- Lemieux, L.I.; Rahal, S.S.; Kennedy, C.R. PGE2 reduces arachidonic acid release in murine podocytes: Evidence for an autocrine feedback loop. Am. J. Physiol. Cell Physiol. 2003, 284, C302–C309.
- Yang, L.; Pan, Y.; Wu, Y.; Lin, S.; Dai, B.; Chen, H.; Wan, J. Excessive arachidonic acid induced actin bunching remodeling and podocyte injury via a PKA-c-Abl dependent pathway. Exp. Cell Res. 2020, 388, 111808.
- Pan, Y.; Wan, J.; Liu, Y.; Yang, Q.; Liang, W.; Singhal, P.C.; Saleem, M.A.; Ding, G. sPLA2 IB induces human podocyte apoptosis via the M-type phospholipase A2 receptor. Sci. Rep. 2014, 4, 6660.
- Djudjaj, S.; Boor, P. Cellular and molecular mechanisms of kidney fibrosis. Mol. Asp. Med. 2019, 65, 16–36.
- Duffield, J.S. Cellular and molecular mechanisms in kidney fibrosis. J. Clin. Investig. 2014, 124, 2299–2306.
- Priante, G.; Musacchio, E.; Valvason, C.; Baggio, B. EPA and DHA suppress AngII- and arachidonic acid-induced expression of profibrotic genes in human mesangial cells. J. Nephrol. 2009, 22, 137–143.
- Zimpelmann, J.; Burns, K.D. Angiotensin-(1-7) activates growth-stimulatory pathways in human mesangial cells. Am. J. Physiol. Ren. Physiol. 2009, 296, F337–F346.
- Zhang, C.; Booz, G.W.; Yu, Q.; He, X.; Wang, S.; Fan, F. Conflicting roles of 20-HETE in hypertension and renal end organ damage. Eur. J. Pharmacol. 2018, 833, 190–200.
- Cheng, J.; Garcia, V.; Ding, Y.; Wu, C.C.; Thakar, K.; Falck, J.R.; Ramu, E.; Schwartzman, M.L. Induction of angiotensin-converting enzyme and activation of the renin-angiotensin system contribute to 20-hydroxyeicosatetraenoic acid-mediated endothelial dysfunction. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1917–1924.
- Balakumar, P.; Sambathkumar, R.; Mahadevan, N.; Muhsinah, A.B.; Alsayari, A.; Venkateswaramurthy, N.; Jagadeesh, G. A potential role of the renin-angiotensin-aldosterone system in epithelial-to-mesenchymal transition-induced renal abnormalities: Mechanisms and therapeutic implications. Pharmacol. Res. 2019, 146, 104314.
- Shim, J.H. Prostaglandin E2 Induces Skin Aging via E-Prostanoid 1 in Normal Human Dermal Fibroblasts. Int. J. Mol. Sci. 2019, 20, 5555.
- Lausada, N.; de Gómez Dumm, I.N.; Raimondi, J.C.; de Alaniz, M.J. Effect of cyclosporine and sirolimus on fatty acid desaturase activities in cultured HEPG2 cells. Transplant. Proc. 2009, 41, 1865–1870.
- Whisler, R.L.; Lindsey, J.A.; Proctor, K.V.; Morisaki, N.; Cornwell, D.G. Characteristics of cyclosporine induction of increased prostaglandin levels from human peripheral blood monocytes. Transplantation 1984, 38, 377–381.
- Nielsen, O.H.; Bukhave, K.; Ahnfelt-Rønne, I.; Elmgreen, J. Arachidonic acid metabolism in human neutrophils: Lack of effect of cyclosporine A. Int. J. Immunopharmacol. 1986, 8, 419–426.
- Sipka, S.; Szücs, K.; Szántó, S.; Kovács, I.; Lakos, G.; Antal-Szalmás, P.; Szegedi, G.; Gergely, P. Inhibition of calcineurin activity and protection against cyclosporine A induced cytotoxicity by prednisolone sodium succinate in human peripheral mononuclear cells. Immunopharmacology 2000, 48, 87–92.
- Wondimu, B.; Modéer, T. Cyclosporin A upregulates prostaglandin E2 production in human gingival fibroblasts challenged with tumor necrosis factor alpha in vitro. J. Oral Pathol. Med. 1997, 26, 11–16.
- Smith, H.E.; Jones, J.P., 3rd; Kalhorn, T.F.; Farin, F.M.; Stapleton, P.L.; Davis, C.L.; Perkins, J.D.; Blough, D.K.; Hebert, M.F.; Thummel, K.E.; et al. Role of cytochrome P450 2C8 and 2J2 genotypes in calcineurin inhibitor-induced chronic kidney disease. Pharmacogenet. Genom. 2008, 18, 943–953.
- Croxtall, J.D.; Paul-Clark, M.; Van Hal, P.T. Differential modulation of glucocorticoid action by FK506 in A549 cells. Biochem. J. 2003, 376 Pt 1, 285–290.
- Mazzocchi, A.; Agostoni, C. Long-Chain ω-3 Polyunsaturated Fatty Acids: Do Genetic Steps Match Metabolic Needs? J. Nutr. 2019, 149, 1690–1691.
- Simopoulos, A.P. Evolutionary aspects of diet: The omega-6/omega-3 ratio and the brain. Mol. Neurobiol. 2011, 44, 203–215.
- Nakamura, M.T.; Nara, T.Y. Structure, function, and dietary regulation of delta6, delta5, and delta9 desaturases. Annu. Rev. Nutr. 2004, 24, 345–376.
- Ameur, S.; Enroth, S.; Johansson, A.; Zaboli, G.; Igl, W.; Johansson, A.C.V.; Rivas, M.A.; Daly, M.J.; Schmitz, G.; Hicks, A.A.; et al. Genetic adaptation of fatty acid metabolism: A human specific haplotype increasing the biosyntesis of long-chain omega-3 and omega -6 fatty acid. Am. J. Hum. Genet. 2012, 90, 809–820.
- Marangoni, F.; Agostoni, C.; Borghi, C.; Catapano, A.L.; Cena, H.; Ghiselli, A.; La Vecchia, C.; Lercker, G.; Manzato, E.; Pirillo, A.; et al. Dietary linoleic acid and human health: Focus on cardiovascular and cardiometabolic effects. Atherosclerosis 2020, 292, 90–98.
- Ramsden, C.E.; Zamora, D.; Makriyannis, A.; Wood, J.T.; Mann, J.D.; Faurot, K.R.; MacIntosh, B.A.; Majchrzak-Hong, S.F.; Gross, J.R.; Courville, A.B.; et al. Diet-induced changes in n-3- and n-6-derived endocannabinoids and reductions in headache pain and psychological distress. J. Pain 2015, 16, 707–716.
- Forsyth, S.; Gautier, S.; Salem, N., Jr. Global Estimates of Dietary Intake of Docosahexaenoic Acid and Arachidonic Acid in Developing and Developed Countries. Ann. Nutr. Metab. 2016, 68, 258–267.