Oximes have been studied for decades because of their significant roles as acetylcholinesterase reactivators. Over the last twenty years, a large number of oximes have been reported with useful pharmaceutical properties, including compounds with antibacterial, anticancer, anti-arthritis, and anti-stroke activities. Many oximes are kinase inhibitors and have been shown to inhibit over 40 different kinases, including AMP-activated protein kinase (AMPK), phosphatidylinositol 3-kinase (PI3K), cyclin-dependent kinase (CDK), serine/threonine kinases glycogen synthase kinase 3 α/β (GSK-3α/β), Aurora A, B-Raf, Chk1, death-associated protein-kinase-related 2 (DRAK2), phosphorylase kinase (PhK), serum and glucocorticoid-regulated kinase (SGK), Janus tyrosine kinase (JAK), and multiple receptor and non-receptor tyrosine kinases. Some oximes are inhibitors of lipoxygenase 5, human neutrophil elastase, and proteinase 3. The oxime group contains two H-bond acceptors (nitrogen and oxygen atoms) and one H-bond donor (OH group), versus only one H-bond acceptor present in carbonyl groups. This feature, together with the high polarity of oxime groups, may lead to a significantly different mode of interaction with receptor binding sites compared to corresponding carbonyl compounds, despite small changes in the total size and shape of the compound. In addition, oximes can generate nitric oxide.
1. Introduction
Oxime compounds have been investigated for decades because of their significant roles as acetylcholinesterase reactivators and their use as therapeutics for a number of diseases
[1][2][3]. Metabolites of various oximes have also been identified in plants as intermediates in biosynthesis and can facilitate a range of processes important for plant growth and development (for review
[4]). Since amidoximes were found to be synthetic antimicrobial agents
[5], oximes with different scaffolds have been developed for the treatment of bacterial infections, including tuberculosis
[6][7][8][9][10]. Oximes have also been reported to exhibit a wide range of biological activities, such as anti-inflammatory
[11][12][13][14][15] and anti-human immunodeficiency (HIV) agents that can inhibit HIV protease
[16][17]. Indeed, the anti-inflammatory activity of some oximes has been reported to be comparable to standard anti-inflammatory drugs, such as indomethacin, diclofenac, and dexamethasone
[18][19][20]. On the other hand, the introduction of an oxime group into an appropriate chemical backbone is a reasonable approach for the preparation of cytotoxic agents, and many oxime derivatives have been reported to have therapeutic activity for cancer
[2][21][22][23][24][25][26][27] and neurodegenerative disorders
[28][29][30].
The introduction of oxime groups has been reported to increase the biological activity of several natural compounds (Figure 1). For example, oxime derivatives of gossypol, a natural phenol derived from the cotton plant, exhibit antiviral, insecticidal, and fungicidal activity
[31]. Another example is psammaplin A analog, the free oxime group which was responsible for high anticancer activity
[32]. Moreover, oxime derivatives of radicicol, a macrocyclic antifungal antibiotic, showed higher inhibitory activity toward Src tyrosine kinase and anticancer activity in comparison with the parent compound
[33][34]. Similarly, the oxime modifications made on the biflorin structure led to an increase in antibacterial potential
[7]. Acylated oximes derived from triterpenes have shown cytotoxic or antiproliferative activity against many lines of cancer cells
[35]. The biological activity of several indirubin oxime derivatives is much higher than that of the plant alkaloid indirubin
[36][37]. Finally, we recently reported that the oxime derivative of the natural alkaloid tryptanthrin is a c-Jun N-terminal kinase (JNK) inhibitor
[38] (Figure 1).
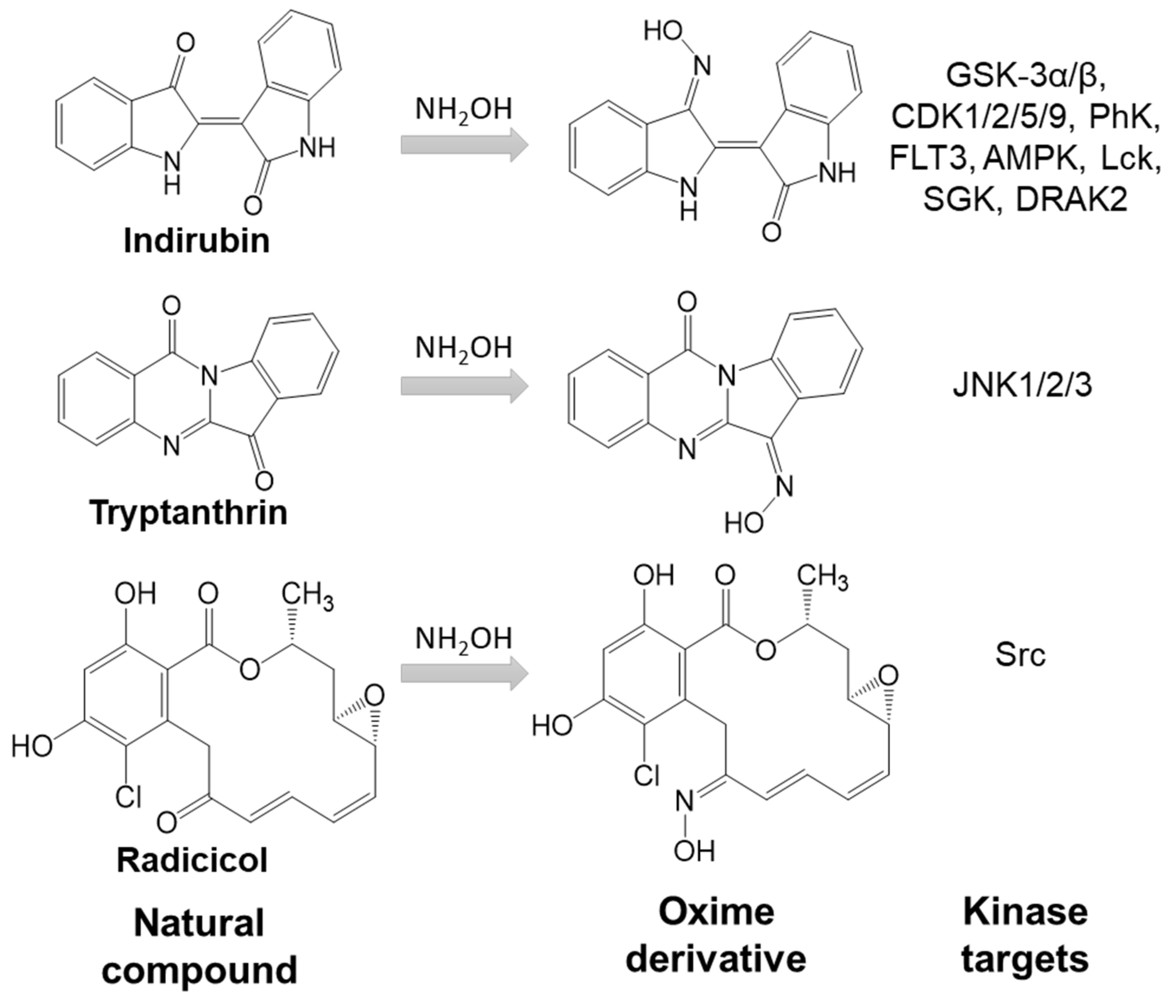
Figure 1. Introduction of oxime groups increases kinase inhibitory activity of natural compounds.
Oximes have been used in the design of various kinase inhibitors, including phosphatidyl inositol 3-kinase (PI3K) inhibitors
[39], phosphorylase kinase (PhK)
[40], and JNK
[38][41]. For example, indirubin oximes are of interest because of their high affinity binding to the ATP-binding site of protein kinases involved in tumorigenesis, e.g., cyclin-dependent kinases (CDK), glycogen synthase kinase (GSK) 3β, vascular endothelial growth factor receptor 2 (VEGFR-2), c-Src, and casein kinase 2 (CK2)
[42][43][44][45][46][47][48]. Many of these kinases are molecular targets for compounds with anticancer activity.
2. Oximes with Non-kinase Targets
While most of the identified oxime targets have been various kinases, there are some oximes that also have non-kinase targets of action. These targets include 5-lipoxygenase (5-LO), proteases, phosphodiesterase, chemokine receptors, growth factor receptors, and various channels (Table 1). For example, several indirubin oximes, such as compounds
1 and
11, have been reported to inhibit 5-LO
[49], which is required for leukotriene synthesis. Replacement of the 3′-oxime in
1 by a keto group, 3′-methoxime or acetoxime resulted in loss of 5-LO inhibitory activity, indicating that a free oxime moiety in the 3′-position and a hydrogen in position N1 are required for effective inhibitory activity
[49]. Additionally, newer derivatives of oleanolic acid oxime, and particularly their conjugates with acetylsalicylic acid, have been shown to downregulate the expression of cyclooxygenase 2 (COX-2) in human hepatoma HepG2 cells by modulating NF-κB signaling
[50]. A reduction in COX-2 leads to reduced prostaglandin synthesis, which also inhibits inflammation in a similar fashion to other nonsteroidal anti-inflammatory drugs (NSAIDs).
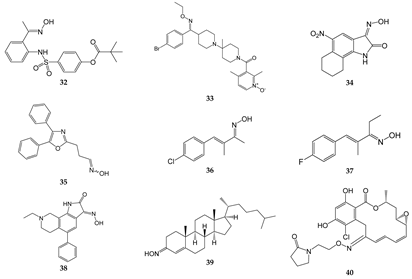
Table 1. Chemical structures of oximes with non-kinase targets and mechanisms of action.
| Compound |
Molecular Target/Mechanism |
Ref. |
| 32 |
Dual inhibitor of HNE and Pr3 |
[15] |
| 33 |
CCR5 antagonist |
[51][52] |
| 34 |
GluR6 antagonist, amelioration of inflammatory hyperalgesia |
[53][54] |
| 35 |
TRPA1 and TRPV1 antagonist |
[14] |
| 36 |
TRPA1 antagonist |
[55][56] |
| 37 |
TRPA1 antagonist |
[55][56] |
| 38 |
ASIC blocker, attenuation of pathophysiological nociceptive behaviors in CFA-inflamed and CCI rats |
[57] |
| 39 |
Binds directly to two components of the mitochondrial permeability pore, the VDAC, and translocator protein; inhibits MPTP opening |
[58] |
| 40 |
Binds to Hsp90 and provides a significant decrease in HIF-1α expression |
[59] |
3. Conclusions and Perspectives
Oxime groups have been successfully introduced into a large number of therapeutic leads for the development of kinase inhibitors with anticancer and anti-inflammatory activities. The kinase selectivity of oximes does not appear to be due to the oxime group. Rather, selectivity seems to be due to the scaffold of the molecule, since some oximes are highly selective (e.g., JNK inhibitors
30 and
31 [38][41]), while others, such as indirubin, have a wide spectrum of kinase targets. In this regard, compounds
30 and
31 are of particular interest as candidates for the development of new anti-inflammatory drugs, since they are highly selective for JNKs.
While the presence of a terminal oxime group is necessary for the activity of these compounds, the oxime group also offers a significant advantage in drug design versus carbonyl groups because of the presence of two H-bond acceptors (N and O atoms) and one donor (OH group). Additionally, the metabolism of oximes can lead to the release of NO, which may also be therapeutically beneficial
[60]. The important role of the oxime group is supported by docking results revealing direct participation of oxime moiety in interactions with kinase binding sites. On the other hand, there has been some concern regarding the development of new drugs based on oxime derivatives. For example, a disadvantage of compound
11 and other indirubin derivatives is the high affinity of indirubin for ATP-binding pockets and the high degree of similarity between ATP cavities within the serine/threonine and tyrosine kinases, leading to multi-targeting. However, single molecules targeting two (or three) kinases is considered less problematic for current pharmaceutical development, and
11 is considered to have significant potential as a therapeutic for treatment of inflammatory and degenerative diseases. One major unsolved issue related to oxime derivatives is their unfavorable physicochemical properties, including poor solubility and membrane permeability, which results in low plasma bioavailability and a short half-life that limits their suitability as drugs
[61][62]. However, compounds
1 and
30 can apparently cross the BBB easily, suggesting that these oximes might be useful for treating brain disorders. New approaches are being developed to improve oxime PK/PD parameters
[63][64][65]. For example, complexing oxime molecules into a dendrimer carrier has been proposed as a strategy to extend their plasma duration through a mechanism of release kinetics, so that loaded drug molecules are released over a longer half-life. Choi et al.
[65] demonstrated that drug-dendrimer complexes form in a specific manner, wherein each oxime molecule interacts through electrostatic attraction with the primary amine terminated at the peripheral branch of the dendrimer
[65]. The importance of the oxime group in kinase binding suggests that additional introduction of this group in the structures of known kinase inhibitors could improve their potency. In addition, oximes with non-kinase targets could be screening toward a broad kinase panel for identification of novel kinase inhibitors.
It is important to note that most of the oximes reviewed here were discovered during compound optimization and not high-throughput screening (HTS). In addition, most of these compounds were characterized in cell-free enzymatic systems and supported in independent test systems. Although compound
30 was originally discovered using HTS in a cell-based assay, the target of this compound was verified using multiple enzymatic assays, cell-based assays, structure–activity relationship (SAR) analysis, and animal experiments. Based on this compound and the absolute requirement for the oxime group in JNK inhibitory activity, we also developed compound
31, which was also validated in cell- and enzyme-based assays and in animal experiments. Thus, it is unlikely that these compounds or the oximes reviewed here are pan assay interference compounds (PAINS)
[66][67]. Nevertheless, this is an important consideration in small molecule screening and will need to be addressed as oximes are developed for new therapeutics.
 +1 credit
+1 credit