Oximes have been studied for decades because of their significant roles as acetylcholinesterase reactivators. Over the last twenty years, a large number of oximes have been reported with useful pharmaceutical properties, including compounds with antibacterial, anticancer, anti-arthritis, and anti-stroke activities. Many oximes are kinase inhibitors and have been shown to inhibit over 40 different kinases, including AMP-activated protein kinase (AMPK), phosphatidylinositol 3-kinase (PI3K), cyclin-dependent kinase (CDK), serine/threonine kinases glycogen synthase kinase 3 α/β (GSK-3α/β), Aurora A, B-Raf, Chk1, death-associated protein-kinase-related 2 (DRAK2), phosphorylase kinase (PhK), serum and glucocorticoid-regulated kinase (SGK), Janus tyrosine kinase (JAK), and multiple receptor and non-receptor tyrosine kinases. Some oximes are inhibitors of lipoxygenase 5, human neutrophil elastase, and proteinase 3. The oxime group contains two H-bond acceptors (nitrogen and oxygen atoms) and one H-bond donor (OH group), versus only one H-bond acceptor present in carbonyl groups. This feature, together with the high polarity of oxime groups, may lead to a significantly different mode of interaction with receptor binding sites compared to corresponding carbonyl compounds, despite small changes in the total size and shape of the compound. In addition, oximes can generate nitric oxide.
- oxime
- kinase inhibitor
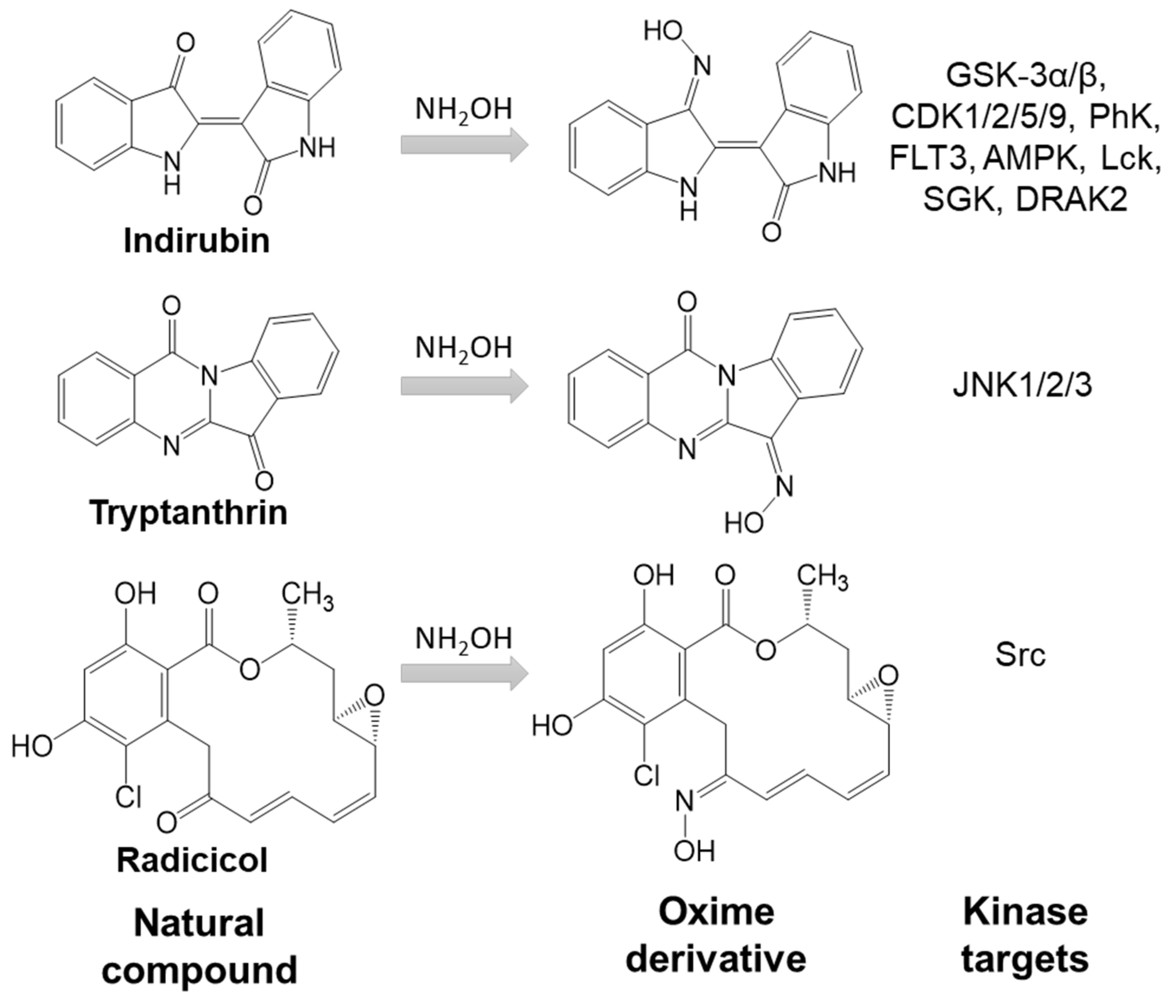
- indirubin
- nitric oxide
- molecular modeling
- inflammation
- cancer
1. Introduction
2. Oximes with Non-kinase Targets
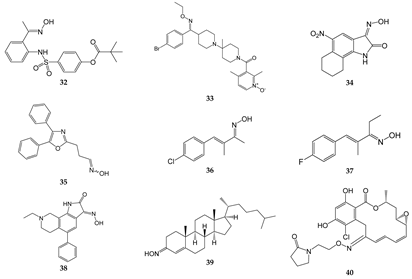
| Compound | Molecular Target/Mechanism | Ref. |
|---|---|---|
| 32 | Dual inhibitor of HNE and Pr3 | [15] |
| 33 | CCR5 antagonist | [51][52] |
| 34 | GluR6 antagonist, amelioration of inflammatory hyperalgesia | [53][54] |
| 35 | TRPA1 and TRPV1 antagonist | [14] |
| 36 | TRPA1 antagonist | [55][56] |
| 37 | TRPA1 antagonist | [55][56] |
| 38 | ASIC blocker, attenuation of pathophysiological nociceptive behaviors in CFA-inflamed and CCI rats | [57] |
| 39 | Binds directly to two components of the mitochondrial permeability pore, the VDAC, and translocator protein; inhibits MPTP opening | [58] |
| 40 | Binds to Hsp90 and provides a significant decrease in HIF-1α expression | [59] |
3. Conclusions and Perspectives
This entry is adapted from the peer-reviewed paper 10.3390/biom11060777
References
- Musilek, K.; Dolezal, M.; Gunn-Moore, F.; Kuca, K. Design, evaluation and structure-activity relationship studies of the AChE reactivators against organophosphorus pesticides. Med. Res. Rev. 2011, 31, 548–575.
- Canario, C.; Silvestre, S.; Falcao, A.; Alves, G. Steroidal oximes: Useful compounds with antitumor activities. Curr. Med. Chem. 2018, 25, 660–686.
- Franjesevic, A.J.; Sillart, S.B.; Beck, J.M.; Vyas, S.; Callam, C.S.; Hadad, C.M. Resurrection and reactivation of acetylcholinesterase and butyrylcholinesterase. Chemistry 2019, 25, 5337–5371.
- Sorensen, M.; Neilson, E.H.J.; Moller, B.L. Oximes: Unrecognized chameleons in general and specialized plant metabolism. Mol. Plant 2018, 11, 95–117.
- Fuller, A.T. Antibacterial action of some aromatic amines, amidines, amidoximes, guanidines and diguanides. Biochem. J. 1947, 41, 403–408.
- Fylaktakidou, K.C.; Hadjipavlou-Litina, D.J.; Litinas, K.E.; Varella, E.A.; Nicolaides, D.N. Recent developments in the chemistry and in the biological applications of amidoximes. Curr. Pharm. Des. 2008, 14, 1001–1047.
- Souza, L.G.D.; Almeida, M.C.S.; Lemos, T.L.G.; Ribeiro, P.R.V.; de Brito, E.S.; Silva, V.L.M.; Silva, A.M.S.; Braz, R.; Costa, J.G.M.; Rodrigues, F.F.G.; et al. Synthesis, antibacterial and cytotoxic activities of new biflorin-based hydrazones and oximes. Bioorg. Med. Chem. Lett. 2016, 26, 435–439.
- Reddy, D.S.; Kongot, M.; Netalkar, S.P.; Kurjogi, M.M.; Kumar, R.; Avecilla, F.; Kumar, A. Synthesis and evaluation of novel coumarin-oxime ethers as potential anti-tubercular agents: Their DNA cleavage ability and BSA interaction study. Eur. J. Med. Chem. 2018, 150, 864–875.
- Hall, J.E.; Kerrigan, J.E.; Ramachandran, K.; Bender, B.C.; Stanko, J.P.; Jones, S.K.; Patrick, D.A.; Tidwell, R.R. Anti-pneumocystis activities of aromatic diamidoxime prodrugs. Antimicrob. Agents Chemother. 1998, 42, 666–674.
- Clement, B.; Burenheide, A.; Rieckert, W.; Schwarz, J. Diacetyldiamidoximeester of pentamidine, a prodrug for treatment of protozoal diseases: Synthesis, in vitro and in vivo biotransformation. ChemMedChem 2006, 1, 1260–1267.
- Li, Q.; Zhang, J.P.; Chen, L.Z.; Wang, J.Q.; Zhou, H.P.; Tang, W.J.; Xue, W.; Liu, X.H. New pentadienone oxime ester derivatives: Synthesis and anti-inflammatory activity. J. Enzym. Inhib. Med. Chem. 2017, 33, 130–138.
- Liu, C.; Tang, X.; Zhang, W.; Li, G.; Chen, Y.; Guo, A.; Hu, C. 6-bromoindirubin-3′-oxime suppresses LPS-induced inflammation via inhibition of the TLR4/NF-κB and TLR4/MAPK signaling pathways. Inflammation 2019, 42, 2192–2204.
- Kwon, Y.J.; Yoon, C.H.; Lee, S.W.; Park, Y.B.; Lee, S.K.; Park, M.C. Inhibition of glycogen synthase kinase-3β suppresses inflammatory responses in rheumatoid arthritis fibroblast-like synoviocytes and collagen-induced arthritis. Jt. Bone Spine 2014, 81, 240–246.
- Payrits, M.; Saghy, E.; Matyus, P.; Czompa, A.; Ludmerczki, R.; Deme, R.; Sandor, Z.; Helyes, Z.; Szoke, E. A novel 3-(4,5-diphenyl-1,3-oxazol-2-yl)propanal oxime compound is a potent transient receptor potential ankyrin 1 and vanilloid 1 (TRPA1 and V1) receptor antagonist. Neuroscience 2016, 324, 151–162.
- Hwang, T.L.; Wang, W.H.; Wang, T.Y.; Yu, H.P.; Hsieh, P.W. Synthesis and pharmacological characterization of 2-aminobenzaldehyde oxime analogs as dual inhibitors of neutrophil elastase and proteinase 3. Bioorg. Med. Chem. 2015, 23, 1123–1134.
- Komai, T.; Yagi, R.; Suzuki-Sunagawa, H.; Ishikawa, Y.; Kasuya, A.; Miyamoto, S.; Handa, H.; Nishigaki, T. Inhibition of HIV-1 protease by oxim derivatives. Biochem. Biophys. Res. Commun. 1997, 230, 557–561.
- Heredia, A.; Davis, C.; Bamba, D.; Le, N.; Gwarzo, M.Y.; Sadowska, M.; Gallo, R.C.; Redfield, R.R. Indirubin-3 ‘-monoxime, a derivative of a chinese antileukemia medicine, inhibits P-TEFb function and HIV-1 replication. AIDS 2005, 19, 2087–2095.
- Chaubal, R.; Mujumdar, A.M.; Misar, A.; Deshpande, V.H.; Deshpande, N.R. Structure-activity relationship study of androstene steroids with respect to local anti-inflammatory activity. Arzneimittelforschung 2006, 56, 394–398.
- Antoniadou-Vyza, E.; Avramidis, N.; Kourounakis, A.; Hadjipetrou, L. Anti-inflammatory properties of new adamantane derivatives. Design, synthesis, and biological evaluation. Arch. Pharm. 1998, 331, 72–78.
- Zeferino-Diaz, R.; Olivera-Castillo, L.; Davalos, A.; Grant, G.; Kantun-Moreno, N.; Rodriguez-Canul, R.; Bernes, S.; Sandoval-Ramirez, J.; Fernandez-Herrera, M.A. 22-oxocholestane oximes as potential anti-inflammatory drug candidates. Eur. J. Med. Chem. 2019, 168, 78–86.
- Shen, S.; Xu, N.; Klamer, G.; Ko, K.H.; Khoo, M.; Ma, D.; Moore, J.; O’Brien, T.A.; Dolnikov, A. Small-molecule inhibitor of glycogen synthase kinase 3β 6-bromoindirubin-3-oxime inhibits hematopoietic regeneration in stem cell recipient mice. Stem. Cells Dev. 2015, 24, 724–736.
- Zhang, X.; Castanotto, D.; Nam, S.; Horne, D.; Stein, C. 6bio enhances oligonucleotide activity in cells: A potential combinatorial anti-androgen receptor therapy in prostate cancer cells. Mol. Ther. 2017, 25, 79–91.
- Qu, H.E.; Huang, R.Z.; Yao, G.Y.; Li, J.L.; Ye, M.Y.; Wang, H.S.; Liu, L. Synthesis and pharmacological evaluation of novel bisindole derivatives bearing oximes moiety: Identification of novel proapoptotic agents. Eur. J. Med. Chem. 2015, 95, 400–415.
- Chiou, C.T.; Lee, W.C.; Liao, J.H.; Cheng, J.J.; Lin, L.C.; Chen, C.Y.; Song, J.S.; Wu, M.H.; Shia, K.S.; Li, W.T. Synthesis and evaluation of 3-ylideneoxindole acetamides as potent anticancer agents. Eur. J. Med. Chem. 2015, 98, 1–12.
- Blazevic, T.; Heiss, E.H.; Atanasov, A.G.; Breuss, J.M.; Dirsch, V.M.; Uhrin, P. Indirubin and indirubin derivatives for counteracting proliferative diseases. Evid. Based Complement. Alternat. Med. 2015, 2015, 654098.
- Xiong, B.; Chen, S.; Zhu, P.; Huang, M.; Gao, W.; Zhu, R.; Qian, J.; Peng, Y.; Zhang, Y.; Dai, H.; et al. Design, synthesis, and biological evaluation of novel thiazolyl substituted bis-pyrazole oxime derivatives with potent antitumor activities by selectively inducing apoptosis and ROS in cancer cells. Med. Chem. 2019, 15, 743–754.
- Galmozzi, E.; Facchetti, F.; La Porta, C.A. Cancer stem cells and therapeutic perspectives. Curr. Med. Chem. 2006, 13, 603–607.
- Avrahami, L.; Farfara, D.; Shaham-Kol, M.; Vassar, R.; Frenkel, D.; Eldar-Finkelman, H. Inhibition of glycogen synthase kinase-3 ameliorates β-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the alzheimer disease mouse model: In vivo and in vitro studies. J. Biol. Chem. 2013, 288, 1295–1306.
- Sathiya Priya, C.; Vidhya, R.; Kalpana, K.; Anuradha, C.V. Indirubin-3′-monoxime prevents aberrant activation of gsk-3beta/nf-kappab and alleviates high fat-high fructose induced abeta-aggregation, gliosis and apoptosis in mice brain. Int. Immunopharmacol. 2019, 70, 396–407.
- Yuskaitis, C.J.; Jope, R.S. Glycogen synthase kinase-3 regulates microglial migration, inflammation, and inflammation-induced neurotoxicity. Cell. Signal. 2009, 21, 264–273.
- Li, L.; Li, Z.; Wang, K.L.; Liu, Y.X.; Li, Y.Q.; Wang, Q.M. Synthesis and antiviral, insecticidal, and fungicidal activities of gossypol derivatives containing alkylimine, oxime or hydrazine moiety. Bioorg. Med. Chem. 2016, 24, 474–483.
- Hong, S.; Shin, Y.; Jung, M.; Ha, M.W.; Park, Y.; Lee, Y.J.; Shin, J.; Oh, K.B.; Lee, S.K.; Park, H.G. Efficient synthesis and biological activity of psammaplin a and its analogues as antitumor agents. Eur. J. Med. Chem. 2015, 96, 218–230.
- Soga, S.; Neckers, L.M.; Schulte, T.W.; Shiotsu, Y.; Akasaka, K.; Narumi, H.; Agatsuma, T.; Ikuina, Y.; Murakata, C.; Tamaoki, T.; et al. KF25706, a novel oxime derivative of radicicol, exhibits in vivo antitumor activity via selective depletion of Hsp90 binding signaling molecules. Cancer Res. 1999, 59, 2931–2938.
- Ikuina, Y.; Amishiro, N.; Miyata, M.; Narumi, H.; Ogawa, H.; Akiyama, T.; Shiotsu, Y.; Akinaga, S.; Murakata, C. Synthesis and antitumor activity of novel O-carbamoylmethyloxime derivatives of radicicol. J. Med. Chem. 2003, 46, 2534–2541.
- Bednarczyk-Cwynar, B.; Zaprutko, L. Recent advances in synthesis and biological activity of triterpenic acylated oximes. Phytochem. Rev. 2015, 14, 203–231.
- Vougogiannopoulou, K.; Skaltsounis, A.L. From tyrian purple to kinase modulators: Naturally halogenated indirubins and synthetic analogues. Planta Med. 2012, 78, 1515–1528.
- Leclerc, S.; Garnier, M.; Hoessel, R.; Marko, D.; Bibb, J.A.; Snyder, G.L.; Greengard, P.; Biernat, J.; Wu, Y.Z.; Mandelkow, E.M.; et al. Indirubins inhibit glycogen synthase kinase-3β and CDK5/p25, two protein kinases involved in abnormal tau phosphorylation in alzheimer’s disease—A property common to most cycline-dependent kinase inhibitors? J. Biol. Chem. 2001, 276, 251–260.
- Schepetkin, I.A.; Khlebnikov, A.I.; Potapov, A.S.; Kovrizhina, A.R.; Matveevskaya, V.V.; Belyanin, M.L.; Atochin, D.N.; Zanoza, S.O.; Gaidarzhy, N.M.; Lyakhov, S.A.; et al. Synthesis, biological evaluation, and molecular modeling of 11H-indeno[1,2-b]quinoxalin-11-one derivatives and tryptanthrin-6-oxime as c-Jun N-terminal kinase inhibitors. Eur. J. Med. Chem. 2019, 161, 179–191.
- Lu, L.; Sha, S.; Wang, K.; Zhang, Y.H.; Liu, Y.D.; Ju, G.D.; Wang, B.; Zhu, H.L. Discovery of chromeno[4,3-c]pyrazol-4(2H)-one containing carbonyl or oxime derivatives as potential, selective inhibitors PI3Kα. Chem. Pharm. Bull. 2016, 64, 1576–1581.
- Begum, J.; Skamnaki, V.T.; Moffatt, C.; Bischler, N.; Sarrou, J.; Skaltsounis, A.L.; Leonidas, D.D.; Oikonomakos, N.G.; Hayes, J.M. An evaluation of indirubin analogues as phosphorylase kinase inhibitors. J. Mol. Graph. Model. 2015, 61, 231–242.
- Schepetkin, I.A.; Kirpotina, L.N.; Khlebnikov, A.I.; Hanks, T.S.; Kochetkova, I.; Pascual, D.W.; Jutila, M.A.; Quinn, M.T. Identification and characterization of a novel class of c-Jun N-terminal kinase inhibitors. Mol. Pharmacol. 2012, 81, 832–845.
- Nam, S.; Scuto, A.; Yang, F.; Chen, W.; Park, S.; Yoo, H.S.; Konig, H.; Bhatia, R.; Cheng, X.; Merz, K.H.; et al. Indirubin derivatives induce apoptosis of chronic myelogenous leukemia cells involving inhibition of STAT5 signaling. Mol. Oncol. 2012, 6, 276–283.
- Hoessel, R.; Leclerc, S.; Endicott, J.A.; Nobel, M.E.; Lawrie, A.; Tunnah, P.; Leost, M.; Damiens, E.; Marie, D.; Marko, D.; et al. Indirubin, the active constituent of a chinese antileukaemia medicine, inhibits cyclin-dependent kinases. Nat. Cell Biol. 1999, 1, 60–67.
- Meijer, L.; Skaltsounis, A.L.; Magiatis, P.; Polychronopoulos, P.; Knockaert, M.; Leost, M.; Ryan, X.P.; Vonica, C.A.; Brivanlou, A.; Dajani, R.; et al. GSK-3-selective inhibitors derived from tyrian purple indirubins. Chem. Biol. 2003, 10, 1255–1266.
- Chan, Y.K.; Kwok, H.H.; Chan, L.S.; Leung, K.S.; Shi, J.; Mak, N.K.; Wong, R.N.; Yue, P.Y. An indirubin derivative, E804, exhibits potent angiosuppressive activity. Biochem. Pharmacol. 2012, 83, 598–607.
- Nam, S.; Buettner, R.; Turkson, J.; Kim, D.; Cheng, J.Q.; Muehlbeyer, S.; Hippe, F.; Vatter, S.; Merz, K.H.; Eisenbrand, G.; et al. Indirubin derivatives inhibit STAT3 signaling and induce apoptosis in human cancer cells. Proc. Natl. Acad. Sci. USA 2005, 102, 5998–6003.
- Nam, S.; Wen, W.; Schroeder, A.; Herrmann, A.; Yu, H.; Cheng, X.; Merz, K.H.; Eisenbrand, G.; Li, H.; Yuan, Y.C.; et al. Dual inhibition of janus and src family kinases by novel indirubin derivative blocks constitutively-activated STAT3 signaling associated with apoptosis of human pancreatic cancer cells. Mol. Oncol. 2013, 7, 369–378.
- Cheng, X.; Merz, K.H.; Vatter, S.; Christ, J.; Wolfl, S.; Eisenbrand, G. 7,7′-diazaindirubin--a small molecule inhibitor of casein kinase 2 in vitro and in cells. Bioorg. Med. Chem. 2014, 22, 247–255.
- Pergola, C.; Gaboriaud-Kolar, N.; Jestadt, N.; Konig, S.; Kritsanida, M.; Schaible, A.M.; Li, H.K.; Garscha, U.; Weinigel, C.; Barz, D.; et al. Indirubin core structure of glycogen synthase kinase-3 inhibitors as novel chemotype for intervention with 5-lipoxygenase. J. Med. Chem. 2014, 57, 3715–3723.
- Krajka-Kuzniak, V.; Bednarczyk-Cwynar, B.; Paluszczak, J.; Szaefer, H.; Narozna, M.; Zaprutko, L.; Baer-Dubowska, W. Oleanolic acid oxime derivatives and their conjugates with aspirin modulate the NF-κB-mediated transcription in HEPG2 hepatoma cells. Bioorg. Chem. 2019, 93, 103326.
- Strizki, J.M.; Xu, S.; Wagner, N.E.; Wojcik, L.; Liu, J.; Hou, Y.; Endres, M.; Palani, A.; Shapiro, S.; Clader, J.W.; et al. SCH-C (SCH 351125), an orally bioavailable, small molecule antagonist of the chemokine receptor CCR5, is a potent inhibitor of HIV-1 infection in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 12718–12723.
- Tsamis, F.; Gavrilov, S.; Kajumo, F.; Seibert, C.; Kuhmann, S.; Ketas, T.; Trkola, A.; Palani, A.; Clader, J.W.; Tagat, J.R.; et al. Analysis of the mechanism by which the small-molecule CCR5 antagonists SCH-351125 and SCH-350581 inhibit human immunodeficiency virus type 1 entry. J. Virol. 2003, 77, 5201–5208.
- Johansen, T.H.; Drejer, J.; Watjen, F.; Nielsen, E.O. A novel non-NMDA receptor antagonist shows selective displacement of low-affinity [H-3] kainate binding. Eur. J. Pharm. Molec. Pharmacol. 1993, 246, 195–204.
- Guo, W.; Zou, S.P.; Tal, M.; Ren, K. Activation of spinal kainate receptors after inflammation: Behavioral hyperalgesia and subunit gene expression. Eur. J. Pharmacol. 2002, 452, 309–318.
- Petrus, M.; Peier, A.M.; Bandell, M.; Hwang, S.W.; Huynh, T.; Olney, N.; Jegla, T.; Patapoutian, A. A role of TRPA1 in mechanical hyperalgesia is revealed by pharmacological inhibition. Mol. Pain 2007, 3, 40.
- McGaraughty, S.; Chu, K.L.; Perner, R.J.; Didomenico, S.; Kort, M.E.; Kym, P.R. TRPA1 modulation of spontaneous and mechanically evoked firing of spinal neurons in uninjured, osteoarthritic, and inflamed rats. Mol. Pain 2010, 6, 14.
- Munro, G.; Christensen, J.K.; Erichsen, H.K.; Dyhring, T.; Demnitz, J.; Dam, E.; Ahring, P.K. NS383 selectively inhibits acid-sensing ion channels containing 1a and 3 subunits to reverse inflammatory and neuropathic hyperalgesia in rats. CNS Neurosci. Ther. 2016, 22, 135–145.
- Bordet, T.; Buisson, B.; Michaud, M.; Drouot, C.; Galea, P.; Delaage, P.; Akentieva, N.P.; Evers, A.S.; Covey, D.F.; Ostuni, M.A.; et al. Identification and characterization of cholest-4-en-3-one, oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. J. Pharmacol. Exp. Ther. 2007, 322, 709–720.
- Kurebayashi, J.; Otsuki, T.; Kurosumi, M.; Soga, S.; Akinaga, S.; Sonoo, H. A radicicol derivative, KF58333, inhibits expression of hypoxia-inducible factor-1α and vascular endothelial growth factor, angiogenesis and growth of human breast cancer xenografts. Jpn. J. Cancer Res. 2001, 92, 1342–1351.
- Lee, H.J.; Lee, J.; Jeong, P.; Choi, J.; Baek, J.; Ahn, S.J.; Moon, Y.; Heo, J.D.; Choi, Y.H.; Chin, Y.W.; et al. Discovery of a FLT3 inhibitor LDD1937 as an anti-leukemic agent for acute myeloid leukemia. Oncotarget 2018, 9, 924–936.
- Gaboriaud-Kolar, N.; Vougogiannopoulou, K.; Skaltsounis, A.L. Indirubin derivatives: A patent review (2010-present). Expert Opin. Ther. Pat. 2015, 25, 583–593.
- Tchoumtchoua, J.; Halabalaki, M.; Gikas, E.; Tsarbopoulos, A.; Fotaki, N.; Liu, L.; Nam, S.; Jove, R.; Skaltsounis, L.A. Preliminary pharmacokinetic study of the anticancer 6BIO in mice using an UHPLC-MS/MS approach. J. Pharm. Biomed. Anal. 2019, 164, 317–325.
- Lorke, D.E.; Kalasz, H.; Petroianu, G.A.; Tekes, K. Entry of oximes into the brain: A review. Curr. Med. Chem. 2008, 15, 743–753.
- Kobrlova, T.; Korabecny, J.; Soukup, O. Current approaches to enhancing oxime reactivator delivery into the brain. Toxicology 2019, 423, 75–83.
- Choi, S.K.; Thomas, T.P.; Leroueil, P.; Kotlyar, A.; Van Der Spek, A.F.; Baker, J.R., Jr. Specific and cooperative interactions between oximes and pamam dendrimers as demonstrated by 1H NMR study. J. Phys. Chem. B 2012, 116, 10387–10397.
- Baell, J.B. Screening-based translation of public research encounters painful problems. ACS Med. Chem. Lett. 2015, 6, 229–234.
- Dahlin, J.L.; Walters, M.A. How to triage PAINS-full research. Assay Drug Dev. Technol. 2016, 14, 168–174.