Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Julien Vignard | + 3917 word(s) | 1735 | 2021-05-14 04:40:54 | | | |
| 2 | Vivi Li | -3 word(s) | 3914 | 2021-06-03 08:08:55 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Vignard, J. Cytolethal Distending Toxin. Encyclopedia. Available online: https://encyclopedia.pub/entry/10414 (accessed on 25 June 2026).
Vignard J. Cytolethal Distending Toxin. Encyclopedia. Available at: https://encyclopedia.pub/entry/10414. Accessed June 25, 2026.
Vignard, Julien. "Cytolethal Distending Toxin" Encyclopedia, https://encyclopedia.pub/entry/10414 (accessed June 25, 2026).
Vignard, J. (2021, June 02). Cytolethal Distending Toxin. In Encyclopedia. https://encyclopedia.pub/entry/10414
Vignard, Julien. "Cytolethal Distending Toxin." Encyclopedia. Web. 02 June, 2021.
Copy Citation
The cytolethal distending toxin (CDT) is produced by many pathogenic Gram-negative bacteria and is considered as a virulence factor. In human cells, CDT exposure leads to a unique cytotoxicity associated with a characteristic cell distension and induces a cell cycle arrest dependent on the DNA damage response (DDR) triggered by DNA double-strand breaks (DSBs). CDT has thus been classified as a cyclomodulin and a genotoxin.
Gram-negative bacteria
cytolethal distending toxin
DNA damage response
double-strand breaks
cell cycle checkpoints
replicative stress
1. Introduction
The cytolethal distending toxin (CDT) was discovered and described as a heat-labile exotoxin, from both Escherichia coli (EcCDT) and Campylobacter jejuni (CjCDT), able to induce the distension and death of eukaryotic cells [1][2]. Later, different bacterial strains obtained from human clinical isolates were shown to produce CDT, including Haemophilus ducreyi (HdCDT) [3], Aggregatibacter actinomycetemcomitans (AaCDT) [4] and enterohepatic Helicobacter cinaedi [5], all being Gram-negative pathogenic bacterial strains. In addition, Salmonella enterica serovar typhi (S. typhi) displays a CDT-like activity without expressing a typical CDT toxin [6]. Finally, CDT is also found in bacteria colonizing animals, such as Helicobacter species in poultry [7], mice and woodchuck [8] (reviewed in [9]). Up to now, no Gram-positive CDT producing bacteria have been characterized.
Globally, eukaryotic cell exposure to CDT leads to a characteristic cytotoxicity associated with a cell distension phenomenon. CDT also induces a cell cycle arrest dependent on the DNA damage response (DDR), triggered by DNA double-strand breaks (DSBs). In addition to CDTs, only a few bacterial genotoxins have been described, among them the E. coli Usp (uropathogenic-specific protein) [10] and colibactin, characterized in extra-intestinal commensal and pathogenic E. coli strains [11]. Regarding the pathological significance, E. coli Usp is associated with urinary tract infection [12], whereas colibactin has been shown to promote colorectal cancer [13]. In this review, we will focus on CDT and briefly present the structural features of CDT and the trafficking of the catalytic moiety to the host cell nucleus. We will then describe the host cell response to CDT intoxication and, finally, discuss the CDT-related DNA damage characteristics.
1.1. CDT-Related Pathogenicity
The CDT toxin has been involved in diseases development and is thus considered as a virulence factor [14][15]. For example, the pathophysiologic role of CDT has been clearly shown in a rat model for C. jejuni, where only a catalytically-active CjCDT induced damage to the epithelial barrier, diarrhea and severe inflammation of the entire gastro-intestinal tract [16][17]. CDT has also been implicated in Helicobacter hepaticus pathogenicity, as the toxin is key in the development of hepatic dysplastic nodules in an immunocompetent mouse model [18]. Finally, in S. typhi, the pathogen responsible for typhoid fever causing more than 200,000 deaths worldwide per year, the role of the CDT-like typhoid toxin has been characterized. In contrast to a catalytic mutant of the toxin (a mutant of the CDT catalytic moiety), the systemic administration of the purified wild-type typhoid toxin in a mouse model induces almost all of the typhoid fever symptoms [19]. Taken together, these data clearly show that the role of CDT in different pathogenic contexts mainly relies on the catalytic activity of the toxin.
1.2. CDT is a Tripartite A-B Exotoxin
The CDT holotoxin is made of three subunits, CdtA, CdtB and CdtC, encoded by three genes organized in one operon [20][21]. The structures of AaCDT and of HdCDT have been determined [22][23], showing that CDT displays an A-B architecture, like many other exotoxins, where CdtB is the catalytic A-subunit. The B-moiety, essential for the holotoxin binding to the host cell membrane, is composed of the CdtA and CdtC subunits. CDT can therefore be classified as an A-B2 exotoxin. To date, the only known exception is the typhoid toxin of S. typhi, in which the CdtB gene is not associated with CdtA and CdtC, but to PltA and PltB [24], encoding, respectively, the pertussis-like toxin A (homologous to the pertussis toxin ADP-ribosyltransferase subunit) and the pertussis-like toxin B (homologous to one of the pertussis B subunits) [25]. The structure of the typhoid toxin has been solved [19] and shown to be an A2–B5 toxin, the B5 regulatory subunit being composed of a pentameric PltB, whereas the A2 catalytic subunit is composed of the StCdtB and PltA proteins, covalently linked by a disulfide bond.
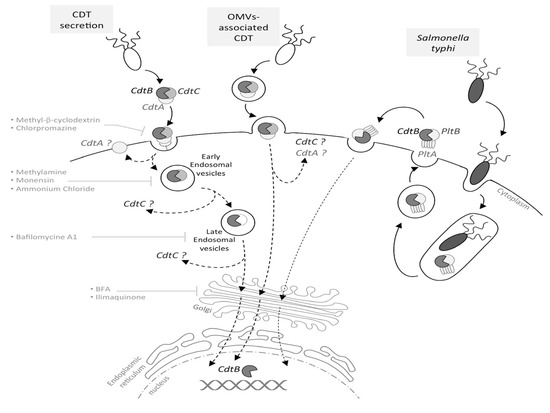
Figure 1. Cytolethal distending toxin (CDT) internalization and trafficking. Depending on the bacteria, CDT may be secreted freely, into outer membrane vesicles (OMVs) or, in the particular case of Salmonella typhi, into intracellular vesicles. In the case of a CDT extracellular secretion, CdtA and CdtC are involved in the toxin binding to the eukaryotic membrane. Once bound, CdtA remains associated with the membrane, while CdtC and CdtB are internalized, with CdtB being relocated to the nucleus by a retrograde transport pathway, via early and late endosomes. This has been demonstrated using inhibitors, such as methyl-β-cyclodextrin, methylamine, bafilomycin A1, BFA, etc. For OMV-secreted CDT, the toxin is internalized into the host cell through the OMV fusion with the eukaryotic membrane. CdtB is relocated to the nucleus by an undescribed pathway (dotted arrow), and the CdtA and CdtC outcome in the cytoplasm is still unknown. The typhoid toxin production requires S. typhi internalization into the host cell; thereafter, the toxin must be secreted to be active. The typhoid toxin interacts with the eukaryotic membrane and is endocytosed, and CdtB is relocated to the nucleus.
CdtA, CdtB and CdtC present a signal sequence and are, therefore, directed to the general secretory pathway, leading to CDT secretion [26][27]. However, CDT may also be released through outer membrane vesicles (OMVs), fusing with the host plasma membrane via lipid rafts [28][29][30]. The S. typhi toxin is once again an exception, as infection studies revealed that the bacterial uptake into host cells triggers the CdtB/PltA/PltB expression, leading to the formation of an intracellular multipartite toxin. Following its production, the typhoid toxin is secreted into the extracellular environment and then interacts, in an autocrine and paracrine way, with the eukaryotic plasma membrane to be internalized and to exert its cytotoxic activity [24], as the inhibition of the typhoid toxin export in the extracellular medium inhibits its effects (Figure 1).
1.3. From CDT Host Cell Binding to CdtB Nuclear Localization
CDT toxins are produced by bacterial strains located in different niches, implying that all CDTs are not secreted in the same microenvironment (different epithelia types, presence of mucus or not, etc.), and this raises questions regarding the cell specificity of the CDT toxins [31]. The CdtB subunit is the most conserved of the subunits among all CDT-producing bacteria strains [32]. On the other hand, significant sequence variability is found between CdtC and CdtA homologs. As CdtA and CdtC subunits are essential to the CDT binding, with CdtB alone not being able to bind at the host cell surface [33][34], some authors hypothesized that this variability may allow CDT to interact specifically with different cell types of the host niches [35]. However, if CdtA and CdtC are involved in the CDT binding to host cells, the nature of the CDT receptor only begins to emerge. Recently, genetic screens identified several eukaryotic cell candidates important for CDT intoxication [36][37]. If different CDT toxins share some host factors as the SGMS1 gene encoding the sphingomyelin synthase 1, some specific host proteins were also found. For example, EcCDT-I specifically requires a putative G protein-coupled receptor, TMEM181, shown to co-immunoprecipitate with CDT [36]. The CDT receptor(s) identification may ultimately provide insights regarding cellular tropism and shed light on host-bacteria interactions.
CDT internalization in host cells seems to occur through receptor-mediated endocytosis [38], where the host receptor bound by the toxin can be considered as a cargo hijacked from its normal cellular function. However, this does not rule out the possibility that CDT enters through other endocytic pathways, such as caveolae, given the importance of cholesterol-rich domains [39][40][41][42], or through cholesterol-independent endocytic pathways [43].
Recently, AaCdtA was shown to stay at the cell surface, whereas AaCdtC was found both on the host cell surface and in the cytosol, AaCdtB being localized at the ER and later at the nucleus [43]. CdtB relocation to the nucleus was also observed after CdtB microinjection into eukaryotic cell cytoplasm [44][45]. Four hours after microinjection, CjCdtB and AaCdtB are located into the nucleus, demonstrating that the nuclear localization of CdtB from the cytoplasm to the nucleus does not require CdtA and CdtC subunits.
After cell entry by endocytosis, HdCdtB seems to follow a retrograde endosome-Golgi traffic to the endoplasmic reticulum (ER) [38], however without requiring the ER-associated degradation (ERAD) pathway usually exploited by ER-translocating toxins [39]. HdCdtB may be directly translocated from the ER to the nucleus, as it could not be seen freely in the intoxicated cells cytosol [46]. Interestingly, the endosomal disruption stops the HdCDT transport, but has no effect on EcCDT-III [47], supporting the hypothesis that different CDT toxins may use different trafficking pathways, possibly through different cargo interactions. Important domains for CdtB nuclear transport have been characterized with green fluorescent protein (GFP) fusions. In the CdtB N-terminal region, an 11-amino acid peptide was found, essential for both nuclear localization and toxin-induced cellular effects [45]. This sequence could be replaced by the nuclear localization signal (NLS) of the SV40 large T-antigen [45], suggesting that CdtB nuclear localization is crucial for CDT cytotoxicity. In addition, two potential NLSs have been identified in the C-terminal part of EcCdtB-II [48], whose deletion produces differential CdtB localizations, suggesting specific functions for these NLS sequences.
To summarize, the CDT trafficking current model involves a stepwise holotoxin disassembly process with: (1) CdtA subunit retention at the plasma membrane, after the CDT toxin binds to a yet unidentified receptor; (2) endocytosis of the CdtB-CdtC complex; and (3) CdtB nuclear localization by a retrograde transport pathway (Figure 1). In addition, CdtC could inhibit the CdtB catalytic activity, as suggested by structural data showing that the N-terminus tail of CdtC occludes the CdtB active site [22]. Hence, once it has entered into the host cell and during its intracellular trafficking, CdtB seems locked-in and has to be released from the CdtC interaction to be active.
2. DNA Damage-Related Cellular Outcomes of CDT Intoxication
Since the discovery of CDT, the cellular response to CDT intoxication has been better and better characterized. CDT induces a cell cycle arrest (at the G2/M transition and, depending on the cell type, at the G1/S transition), accompanied by a cellular distention and, eventually, cell death [49][50][51][52]. These CDT effects showed similarities with those exerted by some DNA damaging agents, such as ionizing radiation (IR) and etoposide, activating similar pathways [50][52][53]. In light of this observation, the apparent sequence homology between CdtB and the endonuclease/exonuclease/phosphatase family encouraged researchers to compare more precisely, among these proteins, CdtB with a well-known nuclease: deoxyribonuclease I (DNase I) [44][54]. As most of the DNase I residues essential for enzymatic activity are strikingly conserved in the different CdtB homologues, potential sites involved in the CdtB nuclease activity have been determined. The corresponding mutants failed to induce any distension, cell death or cell cycle arrest, showing that the CdtB catalytic activity is responsible for the observed cellular effects and that CdtB has a functional homology with DNase I [44][54]. Finally, the CdtB nuclease activity has been demonstrated in vitro by incubating a super-coiled plasmid DNA with the whole CDT holotoxin or with CdtB alone (see below). We reintroduce here the important concepts to study the DNA damage response pathway activation and relate them with the observations made after CDT treatment.
2.1. The CDT-Activated DNA Damage Response
In order to replicate correctly and to maintain the stability of their genetic information, eukaryotic cells display systems for controlling the genome integrity. Cells continuously undergo DNA damage generated by different sources (environment, metabolism, etc.). To survive in the presence of DNA lesions, the cell has developed mechanisms to detect and signal the damage. These interconnected pathways are referred to as the DNA damage response (DDR).
2.1.1. Introduction to the DDR
The DNA damage-related activation of checkpoints is divided into three stages: (i) the recognition of DNA damage by sensor proteins (MRN and Ku complexes, RPA), which rapidly activate specific phosphatidylinositol 3-kinase-related protein kinases (ATM, ATR, DNA-PK) [55]; (ii) the signal amplification by transducing proteins (CHK1, CHK2); which (iii) activates an appropriate cellular response by effectors proteins (p53, CDC25, etc.). This cell response initiates the cell cycle regulation, the activation of DNA repair pathways and, in some cases, cell death pathways [56].
Two key signaling pathways are activated in response to DNA damage: the ATM-CHK2 and the ATR-CHK1 pathways. The ATM-CHK2 pathway is activated with the response to DSB-inducing agents. Following the generation of a DSB, the MRN complex, consisting of MRE11, RAD50 and NBS1, recruits the ATM kinase to the site of injury [57]. Once at the DSB site, ATM is activated by autophosphorylation and phosphorylates hundreds of substrates, including CHK2 and p53. Meanwhile, ATM phosphorylates the H2AX histone (then called γH2AX), several megabases around the DSB site [58], allowing signal amplification. The activated CHK2 phosphorylates various substrates, including p53 and the CDC25 phosphatases family. By contrast, the ATR-CHK1 pathway is activated by the accumulation of single-stranded DNA (ssDNA), particularly during the stalling of replication forks (RFs). Indeed, when replication is blocked by DNA lesions (SSB, DSB, inter-strand and intra-strand crosslinks, base modifications or adducts), DNA polymerase is uncoupled from the replicative helicase, which continues to unwind the DNA and, thus, generates ssDNA. ssDNA is recognized by the RPA protein complex, which protects and stabilizes it, and the accumulation of RPA-coated ssDNA at stalled RFs induces the recruitment of the ATR/ATRIP complex [59]. ATR then phosphorylates the CHK1 transducing protein [60]. ATR can, like ATM, phosphorylate H2AX [61], p53 and many cell cycle regulators (such as CDC25A, CDC25C and Wee1). In conclusion, whatever the activated pathway, the ATR-CHK1 or ATM-CHK2 activation will lead to major protein phosphorylation, involved in various cellular processes, including cell cycle regulation, DNA repair and programmed cell death [62]. However, it has to be underlined that many crosstalk exist between the ATM and ATR pathways (reviewed in [63]). Indeed, although CHK2 is the ATM primary target, ATM can also phosphorylate CHK1. In addition, processing of DSBs during the homologous recombination pathway (HR) generates stretches of ssDNA, leading to the ATR pathway activation [64]. Conversely, prolonged replicative stress can provoke RF collapse [65], resulting in DSB formation and ATM-CHK2 pathway activation (Figure 2). In summary, according to the type of DNA injury, the ATM and ATR pathways can be specifically or sequentially activated, resulting in the cell cycle arrest, the activation of the DNA repair machinery and, potentially, in cell death.
2.1.2. CDT Activates the DNA Damage Response
Many studies have compared the cellular effects of CdtB with DSB induced by IR. The first report deals with the cell cycle arrest induced in response to HdCDT or IR, in HL(human embryonic lung)-fibroblasts and HEp-2 cells [66], and suggested that CDT induces the activation of the DSB-related pathway. To better characterize the CdtB intracellular effects, the activation and the recruitment of different DDR proteins to damaged sites have been studied, demonstrating that CDT exposure recapitulates the different steps of DSB signaling (Figure 3). First, the three subunits of the MRN complex have been shown to form nuclear foci following CDT exposure [67][68][69]. Different studies highlighted an activation of the ATM-dependent pathway, since the ATM protein level was increased and its phosphorylated form accumulated after CDT infection [11][53][70]. Besides, γH2AX foci are formed in response to CDT from various origins [53][67][68][71][72][73][74][75]. γH2AX is the most commonly used DSB biomarker, but can be observed in response to other stresses, such as replicative stress, hypoxia, chromatin remodeling, senescence or cell death [76]. Hence, to strengthen the fact that CDT induces DSB, another DSB marker, 53BP1, has been shown to form foci after CDT treatment [53][73][75]. Finally, ATM activation in response to CDT leads to CHK2 phosphorylation [11][53][70][72][73]. CHK2 phosphorylates different effectors, such as the cell cycle regulators of the phosphatase family, CDC25A and CDC25C. It is well known that ATM and CHK2 activate and stabilize p53, enhancing the transcription of the p21 gene involved in the cell cycle arrest at G1/S [77]. As expected, several studies showed that CDT induces p53 phosphorylation and stabilization, leading to the accumulation of p21 [38][73][78].
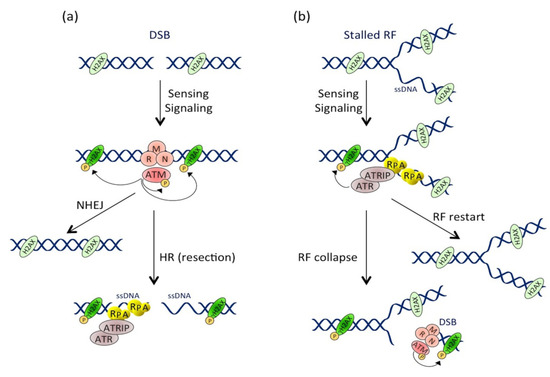
Figure 2. Activation and crosstalk between the ATM and ATR pathways. (a) Pathway activation at the double-stranded break (DSB). DSB formation induces the MRN-dependent ATM recruitment. ATM then phosphorylates numerous substrates, including itself and H2AX near the DSB site. DSB repair occurs through non-homologous end joining (NHEJ) or homologous recombination (HR) mechanisms. During HR, resection of the DSB extremities produces ssDNA stretches that are coated by RPA, leading to ATRIP-dependent ATR recruitment and activation. (b) Pathway activation during replicative stress. Replication fork (RF) stalling induces ssDNA formation that is rapidly coated by RPA. This structure is recognized by ATRIP, which drives the recruitment and activation of ATR. Besides phosphorylating its other substrates, ATR locally phosphorylates H2AX. RF can restart in case of transient stress or collapse after prolonged stresses. RF collapse induces DSB formation and ATM pathway activation, leading to a second and more important wave of H2AX phosphorylation.
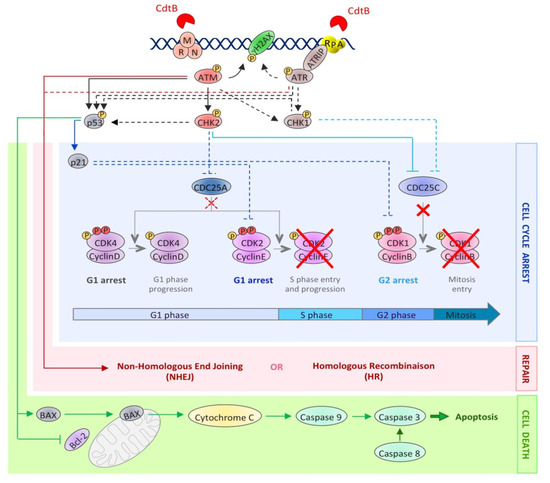
Figure 3. The activation of the DNA damage response upon CDT exposure. This picture depicts the DDR molecular events induced after CDT intoxication. The dotted lines represent well-studied events occurring during the DDR, but not yet demonstrated in the context of CDT exposure. CdtB-induced DNA lesions are detected by sensors, such as the MRN complex and RPA, resulting in the recruitment and the activation of the PI3K related kinases, ATM and ATR. ATM and ATR then phosphorylate hundreds of substrates, including H2AX, CHK1, CHK2 and p53 (black arrows). This signaling cascade results in the regulation of cell cycle modulators (blue lines), through the inhibition of CDC25C phosphatase by CHK2 and, possibly, CHK1. Phosphorylated CDC25C is unable to activate the cyclin B/CDK1 complex (red crosses), essential for mitotic entry. Moreover, the p53-dependent accumulation of p21 blocks cells in G1 by inhibiting the CDK2/cyclin E complex. At the same time, the DSB repair mechanisms (NHEJ and HR) are activated by ATM and ATR (red arrows). However, if the level of DNA lesions is too severe, the process of cell death is initiated (green arrows). Apoptotic cell death can be induced by an intrinsic pathway involving p53 activation, leading to an increase in the BAX level, the sequestration and inactivation of Bcl-2, the mitochondrial release of cytochrome C and caspase 9 activation. Apoptosis can also be induced through the activation of the extrinsic pathway, involving caspase 8 activation. In both cases, this leads to caspase 3 activation and apoptotic cell death.
Few publications studied the activation of the ATR-CHK1 pathway in response to CDT. In 2006, Taieb et al. showed the phosphorylation of ATM, CHK2 and CHK1 proteins following a 24-h treatment with EcCDT. However, in this study, no information on the protein responsible for the CHK1 phosphorylation was given [72]. In a recent publication, the activation of the ATM-CHK2 and ATR-CHK1 pathways, in response to gamma-irradiation or CDT treatment, were compared [53]. Irradiated GM637 fibroblasts present a rapid and transient ATM-dependent CHK1 phosphorylation that precedes a second wave of CHK1 phosphorylation, mediated by ATR, which was not activated during the first 8 h post-irradiation. This delayed ATR-CHK1 pathway activation, as well as the late CHK2 phosphorylation are a consequence of the unrepaired DNA lesions that stall and ultimately collapse the RFs when cells progress through the S-phase. In contrast, following HdCDT treatment, the kinetics of ATR and CHK1 phosphorylation are totally different, while the ATM-CHK2 response is quite similar. Indeed, ATR is found phosphorylated at early time points after CDT exposure, with the same kinetics as CHK1 phosphorylation, which increases over time and is shown to be ATM-independent [53]. These data show that, in contrast to the IR-related DDR response, the ATR-CHK1 pathway is activated early and continuously in response to HdCDT.
2.2. Cellular Effects of the CDT-Induced DNA Damage
2.2.1. Cell Cycle Arrest
The cell-cycle progression is regulated by sequential activation and nuclear relocalization of CDK/Cyclin complexes [79]. At the beginning of the cell cycle, the activation of CDK4/Cyclin D and CDK2/Cyclin E complexes controls the G1-phase progression and entry into S-phase. The CDK2/Cyclin A complex then regulates the S-phase progression. The S-phase completion and G2 transition are coordinated by the activation of the CDK1/Cyclin A complex. Finally mitotic entry depends on the activation and nuclear relocalization of the CDK1/Cyclin B complex. CDK/Cyclin complexes are activated by CDC25-dependent dephosphorylation and can be inhibited by different regulators, like p16, p21, p27 and Wee1 [80].
In response to CDT treatment, CHK2 activation induces the sequestration of CDC25C phosphatase in the cytoplasm, making it unable to activate the CDK1/Cyclin B complex [81]. As a result, the CDK1/Cyclin B complex is hyperphosphorylated [11][52][72][82] and inactive [50], preventing cells from entering the G2-phase (Figure 3). The implication of CDC25C in this process was demonstrated by overexpression of a recombinant CDC25C, which abrogates the CDT-induced G2/M cell cycle arrest [83]. Two other key factors of the cell cycle regulation are p53 and its p21 downstream target. Coherently, the G1/S checkpoint activation in response to CDT relies on the activation of p53 and p21 [84][85][86]. The G1/S checkpoint activation being largely dependent on p53, the variation in the p53 status among the cell types used in the experiments may explain the difference observed for their cell-cycle arrest; most of the p53 negative cells (or with a p53 inactive form) only arresting at G2/M and not at G1/S [68][74][86]. However, p21 induction can also be p53-independent, and a study indeed reported, in response to CDT, a p21 accumulation that was p53-independent [78]. In eukaryotic cells, p21 is known to inhibit many CDK/cyclin complexes, leading to the cell cycle regulation at different steps [87]. CDT intoxication may therefore lead to the cell cycle arrest at the G1/S transition via the CDK2/cyclin E inactivation.
2.2.2. Cell Death or Senescence
Cell cycle arrest is not the only cellular response induced by the DDR. Indeed, the ATM-CHK2 and ATR-CHK1 pathways also activate DNA repair and cell death pathways. Generally, CDT treatment leads to cell death by an apoptotic pathway. CDT-mediated apoptosis has been shown to follow the intrinsic mitochondrial pathway, involving the BAX/Bcl-2 protein [51][74][88], cytochrome C release [74] and caspases activation [74][88][89]. However, few cell lines were shown to use the extrinsic apoptotic pathway through the caspase-8 cleavage [88][89][90]. CDT intoxicated cells die either by apoptosis or necrosis, the latter being perhaps a consequence of abortive mitosis. Hence, some studies demonstrated that in spite of the DDR activation, CDT-treated cells can bypass the G2/M checkpoint, resulting in micronucleation and abortive mitosis [82][91]. Furthermore, endoreplication events have also been documented [49][82]. CDT has been shown to induce an apoptotic response (Figure 3), following the cell cycle arrest, in a broad range of cell lines, with cell death being detectable between two and four days post-intoxication [9]. However, hematopoietic cells seem to activate a rapid apoptotic pathway during the first day of treatment to such an extent that the cell cycle arrest is not even observed [38][51][67][86][92]. When tested under the same conditions, non-hematopoietic cells presented a mild apoptotic response compared to hematopoietic cells, like monocytes and T-cells [93]. Hematopoietic cells therefore show the most dramatic apoptotic response and do not seem to activate the DDR in response to CDT, suggesting a specific cytotoxic mechanism that does not involve CDT-related DNA damage. Hence, such a DNA damage-independent cell death response may rely on the CdtB phosphatase activity [94].
Another possible cellular outcome of the CDT genotoxic effects is the induction of cellular senescence. CDT intoxicated cells express the hallmarks of cellular senescence (i.e., persistently activated DNA damage signaling, enhanced senescence-associated β-galactosidase activity and promyelocytic nuclear compartments expansion) [73]. This is especially important with regard to the senescence-associated secretory phenotype and inflammation [95], as the expression of proinflammatory mediators is observed after CDT infection [18][96]. Finally, this could lead to cytokine-induced bystander genotoxic effects, with a two-fold increase of reactive oxygen species (ROS) observed after a CDT chronic treatment [91].
References
- Johnson, W.M.; Lior, H. Response of chinese hamster ovary cells to a cytolethal distending toxin (CDT) of Escherichia coli and possible misinterpretation as heat-labile (LT) enterotoxin. FEMS Microbiol. Lett. 1987, 43, 19–23.
- Johnson, W.M.; Lior, H. A new heat-labile Cytolethal Distending Toxin (CLDT) produced by campylobacter spp. Microbiol. Pathol. 1988, 4, 115–126.
- Cope, L.D.; Lumbley, S.; Latimer, J.L.; Klesney-Tait, J.; Stevens, M.K.; Johnson, L.S.; Purven, M.; Munson, R.S.; Lagergard, T.; Radolf, J.D.; et al. A diffusible cytotoxin of haemophilus ducreyi. Proc. Natl. Acad. Sci. 1997, 94, 4056–4061.
- Sugai, M.; Kawamoto, T.; Pérès, S.Y.; Ueno, Y.; Komatsuzawa, H.; Fujiwara, T.; Kurihara, H.; Suginaka, H.; Oswald, E. The cell cycle-specific growth-inhibitory factor produced by actinobacillus actinomycetemcomitans is a cytolethal distending toxin. Infect. Immun. 1998, 66, 5008–5019.
- Shen, Z.; Feng, Y.; Rogers, A.B.; Rickman, B.; Whary, M.T.; Xu, S.; Clapp, K.M.; Boutin, S.R.; Fox, J.G. Cytolethal distending toxin promotes helicobacter cinaedi-associated typhlocolitis in interleukin-10-deficient mice. Infect. Immun. 2009, 77, 2508–2516.
- Haghjoo, E.; Galán, J.E. Salmonella typhi encodes a functional cytolethal distending toxin that is delivered into host cells by a bacterial-internalization pathway. Proc. Natl. Acad. Sci. 2004, 101, 4614–4619.
- Stanley, J.; Linton, D.; Burnens, A.P.; Dewhirst, F.E.; On, S.L.W.; Porter, A.; Owen, R.J.; Costas, M. Helicobacter pullorurn sp. nov.-genotype and phenotype of a new species isolated from poultry and from human patients with gastroenteritis. Microbiology 1994, 140, 3441–3449.
- Chien, C.C.; Taylor, N.S.; Ge, Z.; Schauer, D.B.; Young, V.B.; Fox, J.G. Identification of cdtB homologues and cytolethal distending toxin activity in enterohepatic Helicobacter spp. J. Med. Microbiol. 2000, 49, 525–534.
- Jinadasa, R.N.; Bloom, S.E.; Weiss, R.S.; Duhamel, G.E. Cytolethal distending toxin: A conserved bacterial genotoxin that blocks cell cycle progression , leading to apoptosis of a broad range of mammalian cell lineages. Microbiology 2011, 157, 1851–1875.
- Nipic, D.; Podlesek, Z.; Budic, M.; Crnigoj, M.; Zgur-Bertok, D. Escherichia coli uropathogenic-specific protein, usp, is a bacteriocin-like genotoxin. J. Infect. Dis. 2013, 208, 1545–1552.
- Nougayrède, J.; Homburg, S.; Taieb, F.; Boury, M.; Brzuszkiewicz, E.; Gottschalk, G.; Buchrieser, C.; Hacker, J.; Dobrindt, U.; Oswald, E. Escherichia coli induces dna double-strand breaks in eukaryotic cells. Science 2006, 313, 848–851.
- Yamamoto, S.; Nakano, M.; Terai, A.; Yuri, K.; Nakata, K.; Nair, G.B.; Kurazono, H.; Ogawa, O. The presence of the virulence island containing the usp gene in uropathogenic Escherichia coli is associated with urinary tract infection in an experimental mouse model. J. Urol. 2001, 165, 1347–1351.
- Arthur, J.C.; Perez-chanona, E.; Mühlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012, 338, 120–123.
- Finlay, B.B.; Falkow, S. Common themes in microbial pathogenicity revisited. Microbiol. Mol. Biol. Rev. 1997, 61, 136–169.
- Ge, Z.; Schauer, D.B.; Fox, J.G. In vivo virulence properties of bacterial Cytolethal Distending Toxin. Cell. Microbiol. 2008, 10, 1599–1607.
- Jain, D.; Prasad, K.N.; Sinha, S.; Husain, N. Differences in virulence attributes between cytolethal distending toxin positive and negative campylobacter jejuni strains. J. Med. Microbiol. 2008, 57, 267–272.
- Pokkunuri, V.; Pimentel, M.; Morales, W.; Jee, S.R.; Alpern, J.; Weitsman, S.; Marsh, Z.; Low, K.; Hwang, L.; Khoshini, R.; et al. Hole of cytolethal distending toxin in altered stool form and bowel phenotypes in a rat model of post-infectious irritable bowel syndrome. J. Neurogastroenterol. Motil. 2012, 18, 434–442.
- Ge, Z.; Rogers, A.B.; Feng, Y.; Lee, A.; Xu, S.; Taylor, N.S.; Fox, J.G. Bacterial cytolethal distending toxin promotes the development of dysplasia in a model of microbially induced hepatocarcinogenesis. Cell. Microbiol. 2007, 9, 2070–2080.
- Song, J.; Gao, X.; Galán, J.E. Structure and function of the salmonella typhi chimaeric A2B5 typhoid toxin. Nature 2013, 499, 350–354.
- Scott, D.A.; Kaper, J.B. Cloning and sequencing of the genes encoding Escherichia coli cytolethal distending toxin. Infect. Immun. 1994, 62, 244–251.
- Pickett, C.L.; Cottle, D.L.; Pesci, E.C.; Bikah, G. Cloning, sequencing, and expression of the Escherichia coli cytolethal distending toxin genes. Infect. Immun. 1994, 62, 1046–1051.
- Nesić, D.; Hsu, Y.; Stebbins, C.E. Assembly and function of a bacterial genotoxin. Nature 2004, 429, 429–433.
- Yamada, T.; Komoto, J.; Saiki, K.; Konishi, K.; Takusagawa, F. Variation of loop sequence alters stability of cytolethal distending toxin (CDT): Crystal structure of CDT from actinobacillus actinomycetemcomitans. Protein Sci. 2006, 15, 362–372.
- Spanò, S.; Ugalde, J.E.; Galán, J.E. Delivery of a salmonella typhi exotoxin from a host intracellular compartment. Cell Host Microbe 2008, 3, 30–38.
- Kaslow, H.R.; Burns, D.L. Pertussis toxin and target eukaryotic cells: Binding, entry, and activation. FASEB J. 1992, 6, 2685–2690.
- Ueno, Y.; Ohara, M.; Kawamoto, T.; Fujiwara, T.; Komatsuzawa, H.; Oswald, E.; Sugai, M. Biogenesis of the actinobacillus actinomycetemcomitans cytolethal distending toxin holotoxin. Infect. Immun. 2006, 74, 3480–3487.
- Zijnge, V.; Kieselbach, T.; Oscarsson, J. Proteomics of protein secretion by aggregatibacter actinomycetemcomitans. PLoS One 2012, 7, e41662.
- Berlanda Scorza, F.; Doro, F.; Rodríguez-Ortega, M.J.; Stella, M.; Liberatori, S.; Taddei, A.R.; Serino, L.; Gomes Moriel, D.; Nesta, B.; Fontana, M.R.; et al. Proteomics characterization of outer membrane vesicles from the extraintestinal pathogenic Escherichia coli deltatolR IHE3034 mutant. Mol. Cell. Proteomics 2008, 7, 473–485.
- Lindmark, B.; Rompikuntal, P.K.; Vaitkevicius, K.; Song, T.; Mizunoe, Y.; Uhlin, B.E.; Guerry, P.; Wai, S.N. Outer membrane vesicle-mediated release of cytolethal distending toxin (CDT) from campylobacter jejuni. BMC Microbiol. 2009, 9, 220.
- Rompikuntal, P.K.; Thay, B.; Khan, M.K.; Alanko, J.; Penttinen, A.-M.; Asikainen, S.; Wai, S.N.; Oscarsson, J. Perinuclear localization of internalized outer membrane vesicles carrying active cytolethal distending toxin from aggregatibacter actinomycetemcomitans. Infect. Immun. 2012, 80, 31–42.
- Gargi, A.; Reno, M.; Blanke, S.R. Bacterial toxin modulation of the eukaryotic cell cycle: Are all cytolethal distending toxins created equally? Front. Cell. Infect. Microbiol. 2012, 2, 124.
- Hu, X.; Nesic, D.; Stebbins, C.E. Comparative structure-function analysis of cytolethal distending toxins. Proteins 2006, 62, 421–434.
- Lee, R.B.; Hassane, D.C.; Cottle, D.L.; Pickett, C.L. Interactions of campylobacter jejuni cytolethal distending toxin subunits CdtA and CdtC with HeLa cells. Infect. Immun. 2003, 71, 4883–4890.
- Mcsweeney, L.A.; Dreyfus, L.A. Carbohydrate-binding specificity of the Escherichia coli cytolethal distending toxin CdtA-II and CdtC-II subunits. Infect. Immun. 2005, 73, 2051–2060.
- Eshraghi, A.; Maldonado-arocho, F.J.; Gargi, A.; Cardwell, M.M.; Prouty, M.G.; Blanke, S.R.; Bradley, K.A. Cytolethal distending toxin family members are differentially affected by alterations in host glycans and membrane cholesterol. Microbiology 2010, 285, 18199–18207.
- Carette, J.E.; Guimaraes, C.P.; Varadarajan, M.; Park, A.S.; Wuethrich, I.; Godarova, A.; Kotecki, M.; Cochran, B.H.; Spooner, E.; Ploegh, H.L.; et al. Haploid genetic screens in human cells identify host factors used by pathogens. Science 2009, 326, 1231–1235.
- Carette, J.E.; Guimaraes, C.P.; Wuethrich, I.; Blomen, V.A.; Sun, C.; Bell, G.; Yuan, B.; Muellner, M.K.; Nijman, M.; Ploegh, H.L.; et al. Global gene disruption in human cells to assign genes to phenotypes. Nat. Biotechnol. 2011, 29, 542–546.
- Cortes-Bratti, X.; Chaves-Olarte, E.; Lagergård, T.; Thelestam, M. Cellular Internalization of cytolethal distending toxin from haemophilus ducreyi. Infect. Immun. 2000, 68, 6903–6911.
- Guerra, L.; Teter, K.; Lilley, B.N.; Stenerlöw, B.; Holmes, R.K.; Ploegh, H.L.; Sandvig, K.; Thelestam, M.; Frisan, T. Cellular internalization of cytolethal distending toxin: A new end to a known pathway. Cell. Microbiol. 2005, 7, 921–934.
- Boesze-Battaglia, K.; Besack, D.; McKay, T.; Zekavat, A.; Otis, L.; Jordan-Sciutto, K.; Shenker, B.J. Cholesterol-rich membrane microdomains mediate cell cycle arrest induced by actinobacillus actinomycetemcomitans cytolethal-distending toxin. Cell. Microbiol. 2006, 8, 823–836.
- Boesze-Battaglia, K.; Brown, A.; Walker, L.; Besack, D.; Zekavat, A.; Wrenn, S.; Krummenacher, C.; Shenker, B.J. cytolethal distending toxin-induced cell cycle arrest of lymphocytes is dependent upon recognition and binding to cholesterol. J. Biol. Chem. 2009, 284, 10650–10658.
- Lin, C.D.; Lai, C.K.; Lin, Y.H.; Hsieh, J.T.; Sing, Y.T.; Chang, Y.C.; Chen, K.C.; Wang, W.C.; Su, H.L.; Lai, C.H. Cholesterol depletion reduces entry of Campylobacter jejuni cytolethal distending toxin and attenuates intoxication of host cells. Infect. Immun. 2011, 79, 3563–3575.
- Damek-Poprawa, M.; Jang, J.Y.; Volgina, A.; Korostoff, J.; DiRienzo, J.M. Localization of Aggregatibacter actinomycetemcomitans cytolethal distending toxin subunits during intoxication of live cells. Infect. Immun. 2012, 80, 2761–2770.
- Lara-Tejero, M.; Galàn, J.E. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-Like protein. Science 2000, 290, 354–357.
- Nishikubo, S.; Ohara, M.; Ueno, Y.; Ikura, M.; Kurihara, H.; Komatsuzawa, H.; Oswald, E.; Sugai, M. Ann-terminal segment of the active component of the bacterial genotoxin cytolethal distending toxin B (CDTB) directs CDTB into the nucleus. J. Biol. Chem. 2003, 278, 50671–50681.
- Guerra, L.; Nemec, K.N.; Massey, S.; Tatulian, S.A.; Thelestam, M.; Frisan, T.; Teter, K. A novel mode of translocation for cytolethal distending toxin. Biochim. Biophys. Acta 2009, 1793, 489–495.
- Gargi, A.; Tamilselvam, B.; Powers, B.; Prouty, M.G.; Lincecum, T.; Eshraghi, A.; Maldonado-Arocho, F.J.; Wilson, B.A.; Bradley, K.A.; Blanke, S.R. Cellular interactions of the cytolethal distending toxins from Escherichia coli and haemophilus ducreyi. J. Biol. Chem. 2013, 288, 7492–7505.
- McSweeney, L.A.; Dreyfus, L.A. A Nuclear localization of the Escherichia coli cytolethal Distending toxin CdtB subunit. Cell. Microbiol. 2004, 6, 447–458.
- Pérès, S.Y.; Marchès, O.; Daigle, F.; Nougayrède, J.P.; Herault, F.; Tasca, C.; de Rycke, J.; Oswald, E. A new cytolethal distending toxin (CDT) from Escherichia coli producing CNF2 blocks HeLa cell division in G2/M phase. Mol. Microbiol. 1997, 24, 1095–1107.
- Comayras, C.; Tasca, C.; Pe, S.Y.; Oswald, E.; Rycke, J.D.E. Escherichia coli cytolethal distending toxin blocks the HeLa cell cycle at the G2/M transition by preventing cdc2 protein kinase dephosphorylation and activation. Escherichia coli cytolethal distending toxin blocks the HeLa cell cycle at the G 2/M Tr. Infect. Immun. 1997, 65, 5088–5095.
- Ohguchi, M.; Ishisaki, A.; Okahashi, N.; Koide, M.; Koseki, T.; Yamato, K.; Noguchi, T.; Nishihara, T. Actinobacillus actinomycetemcomitans toxin induces both cell cycle arrest in the G2/M phase and apoptosis. Infect. Immun. 1998, 66, 5980–5987.
- Sert, V.; Cans, C.; Tasca, C.; Oswald, E.; Ducommun, B.; de Rycke, J. The bacterial cytolethal distending toxin (CDT) triggers a G2 cell cycle checkpoint in mammalian cells without preliminary induction of DNA strand breaks. Oncogene 1999, 18, 6296–6304.
- Fahrer, J.; Huelsenbeck, J.; Jaurich, H.; Dörsam, B.; Frisan, T.; Eich, M.; Roos, W.P.; Kaina, B.; Fritz, G. Cytolethal distending toxin (CDT) is a radiomimetic agent and induces persistent levels of DNA double-strand breaks in human fibroblasts. DNA Repair (Amst) 2014, 18, 31–43.
- Elwell, C.A.; Dreyfus, L.A. DNase I homologous residues in CdtB are critical for cytolethal distending toxin-mediated cell cycle arrest. Mol. Microbiol. 2000, 37, 952–963.
- Niida, H.; Nakanishi, M. DNA damage checkpoints in mammals. Mutagenesis 2006, 21, 3–9.
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078.
- Lee, J.-H.; Paull, T.T. ATM activation by DNA double-Strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005, 308, 551–554.
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999, 146, 905–916.
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548.
- Zhao, H.; Piwnica-Worms, H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell. Biol. 2001, 21, 4129–4139.
- Ward, I.M.; Chen, J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 2001, 276, 47759–47762.
- Matsuoka, S.; Ballif, B.A; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166.
- Gobbini, E.; Cesena, D.; Galbiati, A.; Lockhart, A.; Longhese, M.P. Interplays between ATM/Tel1 and ATR/Mec1 in sensing and signaling DNA double-strand breaks. DNA Repair (Amst) 2013, 12, 791–799.
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.C.M.; Lukas, J.; Jackson, S.P. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 37–45.
- Feng, W.; di Rienzi, S.C.; Raghuraman, M.K.; Brewer, B.J. Replication stress-induced chromosome breakage is correlated with replication fork progression and is preceded by single-stranded DNA formation. G3 Genes Genomes Genet. 2011, 1, 327–335.
- Cortes-Bratti, X.; Karlsson, C.; Lagergård, T.; Thelestam, M.; Frisan, T. The haemophilus ducreyi cytolethal distending Toxin induces cell cycle arrest and apoptosis via the DNA damage checkpoint pathways. J. Biol. Chem. 2001, 276, 5296–5302.
- Li, L.; Sharipo, A.; Chaves-Olarte, E.; Masucci, M.G.; Levitsky, V.; Thelestam, M.; Frisan, T. The haemophilus ducreyi cytolethal distending toxin activates sensors of DNA damage and repair complexes in proliferating and non-proliferating cells. Cell. Microbiol. 2002, 4, 87–99.
- Hassane, D.C.; Lee, R.B.; Pickett, C.L. Campylobacter jejuni cytolethal distending toxin promotes DNA repair responses in normal human cells. Infect. Immun. 2003, 71, 541–545.
- Guerra, L.; Albihn, A.; Tronnersjö, S.; Yan, Q.; Guidi, R.; Stenerlöw, B.; Sterzenbach, T.; Josenhans, C.; Fox, J.G.; Schauer, D.B.; et al. Myc is required for activation of the ATM-dependent checkpoints in response to DNA damage. PLoS One 2010, 5, e8924.
- Alaoui-El-Azher, M.; Mans, J.J.; Baker, H.V; Chen, C.; Progulske-Fox, A.; Lamont, R.J.; Handfield, M. Role of the ATM-checkpoint kinase 2 pathway in CDT-mediated apoptosis of gingival epithelial cells. PLoS One 2010, 5, e11714.
- Bielaszewska, M.; Sinha, B.; Kuczlus, T.; Karch, H. Cytolethal distending toxin from shiga toxin-producing Escherichia coli O157 causes irreversible G2/M arrest, Inhibition of proliferation, and Death of human endothelial cells. Infect. Immun. 2005, 73, 552–562.
- Taieb, F.; Nougayrède, J.; Watrin, C.; Samba-louaka, A.; Oswald, E. Escherichia coli cyclomodulin Cif induces G 2 arrest of the host cell cycle without activation of the DNA-damage checkpoint-signalling pathway. 2006, 8, 1910–1921.
- Blazkova, H.; Krejcikova, K.; Moudry, P.; Frisan, T.; Hodny, Z.; Bartek, J. Bacterial intoxication evokes cellular senescence with persistent DNA damage and cytokine signalling. J. Cell. Mol. Med. 2010, 14, 357–367.
- Liyanage, N.P.M.; Manthey, K.C.; Dassanayake, R.P.; Kuszynski, C.A.; Oakley, G.G.; Duhamel, G.E. Helicobacter hepaticus cytolethal distending toxin causes cell death in Intestinal epithelial cells via mitochondrial apoptotic pathway. Helicobacter 2010, 15, 98–107.
- Fedor, Y.; Vignard, J.; Nicolau-Travers, M.-L.; Boutet-Robinet, E.; Watrin, C.; Salles, B.; Mirey, G. From single-strand breaks to double-strand breaks during S-phase: A new mode of action of the Escherichia coli cytolethal distending toxin. Cell. Microbiol. 2013, 15, 1–15.
- Vignard, J.; Mirey, G.; Salles, B. Ionizing-radiation induced DNA double-strand breaks: A direct and indirect lighting up. Radiother. Oncol. 2013, 108, 362–369.
- Ahn, J.; Urist, M.; Prives, C. The Chk2 protein kinase. DNA Repair (Amst) 2004, 3, 1039–1047.
- Sato, T.; Koseki, T.; Yamato, K.; Saiki, K.; Konishi, K.; Yoshikawa, M.; Ishikawa, I.; Nishihara, T. p53-Independent Expression of p21 CIP1/WAF1 in plasmacytic cells during G2 cell cycle arrest induced by actinobacillus actinomycetemcomitans cytolethal distending toxin. Infect. Immun. 2002, 70, 528–534.
- Hochegger, H.; Takeda, S.; Hunt, T. Cyclin-dependent kinases and cell-cycle transitions: Does one fit all? Nat. Rev. Mol. Cell Biol. 2008, 9, 910–916.
- Langerak, P.; Russell, P. Regulatory networks integrating cell cycle control with DNA damage checkpoints and double-strand break repair. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 3562–3571.
- Alby, F.; Mazars, R.; de Rycke, J.; Guillou, E.; Baldin, V.; Darbon, J.M.; Ducommun, B. Study of the cytolethal distending toxin (CDT)-activated cell cycle checkpoint. Involvement of the CHK2 kinase. FEBS Lett. 2001, 491, 261–265.
- De Rycke, J.; Sert, V.; Comayras, C.; Tasca, C. Sequence of lethal events in HeLa cells exposed to the G2 blocking cytolethal distending toxin. Eur. J. Cell Biol. 2000, 79, 192–201.
- Escalas, N.; Davezac, N.; de Rycke, J.; Baldin, V.; Mazars, R.; Ducommun, B. Study of the cytolethal distending toxin-induced cell cycle arrest in HeLa cells: Involvement of the CDC25 phosphatase. Exp. Cell Res. 2000, 257, 206–212.
- Bielaszewska, M.; Fell, M.; Greune, L.; Prager, R.; Fruth, A.; Tschäpe, H.; Schmidt, M.A.; Karch, H.; Tscha, H. Characterization of cytolethal distending toxin genes and expression in shiga toxin-producing Escherichia coli strains of non-O157 serogroups. Infect. Immun. 2004, 72, 1812–1816.
- Cortes-Bratti, X.; Frisan, T.; Thelestam, M. The cytolethal distending toxins induce DNA damage and cell cycle arrest. Toxicon 2001, 39, 1729–1736.
- Frisan, T.; Cortes-bratti, X.; Chaves-olarte, E.; Stenerlöw, B.; Thelestam, M.; Rica, U.D.C.; José, S.; Rica, C. The Haemophilus ducreyi cytolethal distending toxin induces DNA double-strand breaks and promotes ATM-dependent activation of RhoA. Cell. Microbiol. 2003, 5, 695–707.
- Abbas, T.; Dutta, A. P21 in Cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414.
- Shenker, B.J.; Hoffmaster, R.H.; Zekavat, A.; Yamaguchi, N.; Lally, E.T.; Demuth, D.R. Induction of apoptosis in human T cells by actinobacillus actinomycetemcomitans cytolethal distending toxin Is a consequence of G2 arrest of the cell cycle. J. Immunol. 2001, 167, 435–441.
- Ohara, M.; Hayashi, T.; Kusunoki, Y.; Miyauchi, M.; Takata, T.; Sugai, M.; Mmun, I.N.I. Caspase-2 and Caspase-7 are involved in cytolethal distending toxin-induced apoptosis in jurkat and MOLT-4 T-cell lines. Infect. Immun. 2004, 72, 871–879.
- Hickey, T.E.; Majam, G.; Guerry, P. Intracellular survival of campylobacter jejuni in human monocytic cells and induction of apoptotic death by cytholethal distending toxin. Infect. Immun. 2005, 73, 5194–5197.
- Guidi, R.; Guerra, L.; Levi, L.; Stenerlöw, B.; Fox, J.G.; Josenhans, C.; Masucci, M.G.; Frisan, T. Chronic exposure to the cytolethal distending toxins of Gram-negative bacteria promotes genomic instability and altered DNA damage response. Cell. Microbiol. 2013, 15, 98–113.
- Gelfanova, V.; Hansen, E.J.; Spinola, S.M. Cytolethal distending toxin of haemophilus ducreyi induces apoptotic death of jurkat T cells. Infect. Immun. 1999, 67, 6394–6402.
- Wising, C.; Azem, J.; Zetterberg, M.; Svensson, L.A.; Ahlman, K.; Lagerga, T. Induction of apoptosis/necrosis in various human cell lineages by haemophilus ducreyi cytolethal distending toxin. Toxicon 2005, 45, 767–776.
- Shenker, B.J.; Dlakic, M.; Walker, L.P.; Besack, D.; Jaffe, E.; LaBelle, E.; Boesze-Battaglia, K. A novel mode of action for a microbial-derivated immunotoxin: The cytolethal distending toxin subunit B exhibits phosphatidylinositol3,4,5-triphosphate phosphatase activity. J. Immunol. 2007, 178, 5099–5108.
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705.
- Guerra, L.; Guidi, R.; Frisan, T. Do bacterial genotoxins contribute to chronic inflammation, genomic instability and tumor progression? FEBS J. 2011, 278, 4577–4588.
More
Information
Subjects:
Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
2 times
(View History)
Update Date:
03 Jun 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No