The cytolethal distending toxin (CDT) is produced by many pathogenic Gram-negative bacteria and is considered as a virulence factor. In human cells, CDT exposure leads to a unique cytotoxicity associated with a characteristic cell distension and induces a cell cycle arrest dependent on the DNA damage response (DDR) triggered by DNA double-strand breaks (DSBs). CDT has thus been classified as a cyclomodulin and a genotoxin.
Cytolethal distending toxins (abbreviated CDTs) are a class of heterotrimeric toxins produced by certain gram-negative bacteria that display DNase activity. These toxins trigger G2/M cell cycle arrest in specific mammalian cell lines, leading to the enlarged or distended cells for which these toxins are named. Affected cells die by apoptosis. Each toxin consists of three distinct subunits named alphabetically in the order that their coding genes appear in the cdt operon. Cytolethal distending toxins are classified as AB toxins, with an active ("A") subunit that directly damages DNA and a binding ("B") subunit that helps the toxin attach to the target cells. CdtB is the active subunit and a homolog to mammalian DNase I, whereas CdtA and CdtC make up the binding subunit. Cytolethal distending toxins are produced by gram-negative pathogenic bacteria from the phylum Pseudomonadota. Many of these bacteria, including Shigella dysenteriae, Haemophilus ducreyi, and Escherichia coli, infect humans. Bacteria that produce CDTs often persistently colonize their host.
1. Introduction
The cytolethal distending toxin (CDT) was discovered and described as a heat-labile exotoxin, from both Escherichia coli (EcCDT) and Campylobacter jejuni (CjCDT), able to induce the distension and death of eukaryotic cells [1][2]. Later, different bacterial strains obtained from human clinical isolates were shown to produce CDT, including Haemophilus ducreyi (HdCDT) [3], Aggregatibacter actinomycetemcomitans (AaCDT) [4] and enterohepatic Helicobacter cinaedi [5], all being Gram-negative pathogenic bacterial strains. In addition, Salmonella enterica serovar typhi (S. typhi) displays a CDT-like activity without expressing a typical CDT toxin [6]. Finally, CDT is also found in bacteria colonizing animals, such as Helicobacter species in poultry [7], mice and woodchuck [8] (reviewed in [9]). Up to now, no Gram-positive CDT producing bacteria have been characterized.
Globally, eukaryotic cell exposure to CDT leads to a characteristic cytotoxicity associated with a cell distension phenomenon. CDT also induces a cell cycle arrest dependent on the DNA damage response (DDR), triggered by DNA double-strand breaks (DSBs). In addition to CDTs, only a few bacterial genotoxins have been described, among them the E. coli Usp (uropathogenic-specific protein) [10] and colibactin, characterized in extra-intestinal commensal and pathogenic E. coli strains [11]. Regarding the pathological significance, E. coli Usp is associated with urinary tract infection [12], whereas colibactin has been shown to promote colorectal cancer [13]. In this review, we will focus on CDT and briefly present the structural features of CDT and the trafficking of the catalytic moiety to the host cell nucleus. We will then describe the host cell response to CDT intoxication and, finally, discuss the CDT-related DNA damage characteristics.
1.2. CDT is a Tripartite A-B Exotoxin
The CDT holotoxin is made of three subunits, CdtA, CdtB and CdtC, encoded by three genes organized in one operon [20][21]. The structures of AaCDT and of HdCDT have been determined [22][23], showing that CDT displays an A-B architecture, like many other exotoxins, where CdtB is the catalytic A-subunit. The B-moiety, essential for the holotoxin binding to the host cell membrane, is composed of the CdtA and CdtC subunits. CDT can therefore be classified as an A-B 1. History
The first recorded observation of a cytolethal-distending toxin was in 1987 in a pathogenic strain in E. coli isolated from a young patient.[3] Later that year, scientists W.M. Johnson and H. Lior published the journal article "Production of Shiga toxin and a cytolethal distending toxin (CLDT) by serogroups of Shigella spp." in Microbiology Letters.[1] The discovery of other bacteria producing CDT toxins continues to this day. In 1994 Scott and Kaper cloned and sequenced a cdt operon from another E. coli strain, publishing in Infection and Immunity.[1][5] The three genes discovered were denoted cdtA, cdtB, and cdtC.[5] In 1997, the first paper of many to show G2/M cell cycle arrest caused by a cytolethal distending toxin was published in Molecular Microbiology.[1] The study focused on another E. coli strain. This paper was followed by a 1999 publication in Infectious Immunity, which demonstrated that H. ducreyi CDT causes cell death via apoptosis. This finding was also confirmed for other cytolethal distending toxins in subsequent studies. The discovery of the homology of cdtB to mammalian DNase I and the current AB model for the toxin were published in early 2000. [2][6] Further research and the publication of crystal structures for the CDT toxins from two different species continues to support this model.[1]
2. Sources
All known cytolethal distending toxins are produced by gram-negative bacteria in the Gammaproteobacteria and Campylobacterota. In several cases, the bacteria producing CDT are human pathogens. Medically important CDT producers include:[1]
- Haemophilus ducreyi (chancroids)
- Aggregatibacter actinomycetemcomitans (periodontitis)
- Escherichia coli (various diseases)
- Shigella dysenteriae (dysentery)
- Salmonella enterica serotype Typhi (typhoid fever)
- Campylobacter upsaliensis (enterocolitis)
- Campylobacter jejuni (enterocolitis)
CDT-producing bacteria are often associated with mucosal linings, such as those in the stomach and intestines, and with persistent infections. The toxins are either secreted freely or associated with the membrane of the producing bacteria.[1]
3. Nomenclature
Individual cytolethal distending toxins are named for the bacterial species that they are isolated from. As of 2011, most scientists have adopted the practice of placing the first letter of both the genus and species in front of the toxin name to reflect its source (i.e., the CDT from Haemaphilus ducreyi is referred to as HdCDT).[1][7] If several subspecies produce different toxins, as in the case of E. coli, Roman numerals may be added after the second letter.[7] Both complete toxins and individual subunits are labeled using this convention. In response to the continued discovery of additional cytolethal distending toxins, a 2011 review has proposed that the toxin names be expanded to include the first three letters of the species (i.e., HducCDT for Haemaphilus ducreyi CDT).[1]
4. Cellular Effects
CDT toxins are genotoxins capable of directly damaging DNA in target cells. They are the only AB-type toxins discovered that display DNase activity, allowing them to introduce breaks into the target cell's DNA.[1][4] In many cell lines including human fibroblasts, epithelial cells, endothelial cells, and keratinocytes, CDTs cause G2/M cell cycle arrest, cytoplasmic distension, and eventual cell death via apoptosis.[1][3][8] Most publications attribute the G2/M cycle arrest to the buildup of irreversible DNA damage from the toxin's DNase activity as the trigger for the G2/M cell cycle arrest, but other research suggests that this model is incomplete.[8] The cytoplasmic distension is a direct result of the G2/M cell cycle arrest. The cell enlarges in preparation for mitosis, but cannot divide to restore its normal size. Aside from classical apoptosis, signs of cellular senescence has also been observed in normal and cancer cell lines (fibroblasts, HeLa and U2-OS) after CDT intoxication[9] In lymphocytes, cell death occurs quickly and is not preceded by significant cytoplasmic distension.[8] The ability of these toxins to effect lymphocytes differently may be advantageous to the bacteria that utilize these toxins, but the mechanism behind this phenomenon is not yet well understood.
5. Toxin Structure
The active, assembled toxin is a tripartite structure with three distinct subunits- CdtA, CdtB, and CdtC. In terms of function, it is an AB toxin. In this context, the CdtB subunit is actually the catalytically active "A" subunit, and the CdtA and CdtC together form the binding "B" subunit, which helps the toxin bind and enter target cells.[6] Some literature refers to the toxin structure as AB
2 exotoxin. To date, the only known exception is the typhoid toxin of S. typhi, in which the CdtB gene is not associated with CdtA and CdtC, but to PltA and PltB [24], encoding, respectively, the pertussis-like toxin A (homologous to the pertussis toxin ADP-ribosyltransferase subunit) and the pertussis-like toxin B (homologous to one of the pertussis B subunits) [25]. The structure of the typhoid toxin has been solved [19] and shown to be an A2–B5 toxin, the B5 regulatory subunit being composed of a pentameric PltB, whereas the A2 catalytic subunit is composed of the StCdtB and PltA proteins, covalently linked by a disulfide bond.
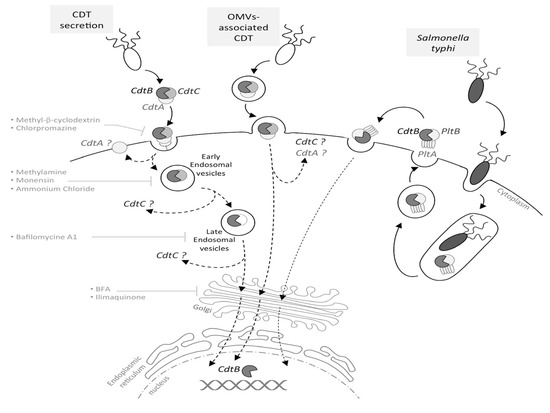
Figure 1. Cytolethal distending toxin (CDT) internalization and trafficking. Depending on the bacteria, CDT may be secreted freely, into outer membrane vesicles (OMVs) or, in the particular case of Salmonella typhi, into intracellular vesicles. In the case of a CDT extracellular secretion, CdtA and CdtC are involved in the toxin binding to the eukaryotic membrane. Once bound, CdtA remains associated with the membrane, while CdtC and CdtB are internalized, with CdtB being relocated to the nucleus by a retrograde transport pathway, via early and late endosomes. This has been demonstrated using inhibitors, such as methyl-β-cyclodextrin, methylamine, bafilomycin A1, BFA, etc. For OMV-secreted CDT, the toxin is internalized into the host cell through the OMV fusion with the eukaryotic membrane. CdtB is relocated to the nucleus by an undescribed pathway (dotted arrow), and the CdtA and CdtC outcome in the cytoplasm is still unknown. The typhoid toxin production requires S. typhi internalization into the host cell; thereafter, the toxin must be secreted to be active. The typhoid toxin interacts with the eukaryotic membrane and is endocytosed, and CdtB is relocated to the nucleus.
CdtA, CdtB and CdtC present a signal sequence and are, therefore, directed to the general secretory pathway, leading to CDT secretion [26][27]. However, CDT may also be released through outer membrane vesicles (OMVs), fusing with the host plasma membrane via lipid rafts [28][29][30]. The S. typhi toxin is once again an exception, as infection studies revealed that the bacterial uptake into host cells triggers the CdtB/PltA/PltB expression, leading to the formation of an intracellular multipartite toxin. Following its production, the typhoid toxin is secreted into the extracellular environment and then interacts, in an autocrine and paracrine way, with the eukaryotic plasma membrane to be internalized and to exert its cytotoxic activity [24], as the inhibition of the typhoid toxin export in the extracellular medium inhibits its effects ().
1.3. From CDT Host Cell Binding to CdtB Nuclear Localization
CDT toxins are produced by bacterial strains located in different niches, implying that all CDTs are not secreted in the same microenvironment (different epithelia types, presence of mucus or not, etc.), and this raises questions regarding the cell specificity of the CDT toxins [31]. The CdtB subunit is the most conserved of the subunits among all CDT-producing bacteria strains [32]. On the other hand, significant sequence variability is found between CdtC and CdtA homologs. As CdtA and CdtC subunits are essential to the CDT binding, with CdtB alone not being able to bind at the host cell surface [33][34], some authors hypothesized that this variability may allow CDT to interact specifically with different cell types of the host niches [35]. However, if CdtA and CdtC are involved in the CDT binding to host cells, the nature of the CDT receptor only begins to emerge. Recently, genetic screens identified several eukaryotic cell candidates important for CDT intoxication [36][37]. If different CDT toxins share some host factors as the SGMS1 gene encoding the sphingomyelin synthase 1, some specific host proteins were also found. For example, EcCDT-I specifically requires a putative G protein-coupled receptor, TMEM181, shown to co-immunoprecipitate with CDT [36]. The CDT receptor(s) identification may ultimately provide insights regarding cellular tropism and shed light on host-bacteria interactions.
CDT internalization in host cells seems to occur through receptor-mediated endocytosis [38], where the host receptor bound by the toxin can be considered as a cargo hijacked from its normal cellular function. However, this does not rule out the possibility that CDT enters through other endocytic pathways, such as caveolae, given the importance of cholesterol-rich domains [39][40][41][42], or through cholesterol-independent endocytic pathways [43].
Recently, AaCdtA was shown to stay at the cell surface, whereas AaCdtC was found both on the host cell surface and in the cytosol, AaCdtB being localized at the ER and later at the nucleus [43]. CdtB relocation to the nucleus was also observed after CdtB microinjection into eukaryotic cell cytoplasm [44][45]. Four hours after microinjection, CjCdtB and AaCdtB are located into the nucleus, demonstrating that the nuclear localization of CdtB from the cytoplasm to the nucleus does not require CdtA and CdtC subunits.
After cell entry by endocytosis, HdCdtB seems to follow a retrograde endosome-Golgi traffic to the endoplasmic reticulum (ER) [38], however without requiring the ER-associated degradation (ERAD) pathway usually exploited by ER-translocating toxins [39]. HdCdtB may be directly translocated from the ER to the nucleus, as it could not be seen freely in the intoxicated cells cytosol [46]. Interestingly, the endosomal disruption stops the HdCDT transport, but has no effect on EcCDT-III [47], supporting the hypothesis that different CDT toxins may use different trafficking pathways, possibly through different cargo interactions. Important domains for CdtB nuclear transport have been characterized with green fluorescent protein (GFP) fusions. In the CdtB N-terminal region, an 11-amino acid peptide was found, essential for both nuclear localization and toxin-induced cellular effects [45]. This sequence could be replaced by the nuclear localization signal (NLS) of the SV40 large T-antigen [45], suggesting that CdtB nuclear localization is crucial for CDT cytotoxicity. In addition, two potential NLSs have been identified in the C-terminal part of EcCdtB-II [48], whose deletion produces differential CdtB localizations, suggesting specific functions for these NLS sequences.
To summarize, the CDT trafficking current model involves a stepwise holotoxin disassembly process with: (1) CdtA subunit retention at the plasma membrane, after the CDT toxin binds to a yet unidentified receptor; (2) endocytosis of the CdtB-CdtC complex; and (3) CdtB nuclear localization by a retrograde transport pathway (). In addition, CdtC could inhibit the CdtB catalytic activity, as suggested by structural data showing that the N-terminus tail of CdtC occludes the CdtB active site [22]. Hence, once it has entered into the host cell and during its intracellular trafficking, CdtB seems locked-in and has to be released from the CdtC interaction to be active.
2. DNA Damage-Related Cellular Outcomes of CDT Intoxication
Since the discovery of CDT, the cellular response to CDT intoxication has been better and better characterized. CDT induces a cell cycle arrest (at the G2/M transition and, depending on the cell type, at the G1/S transition), accompanied by a cellular distention and, eventually, cell death [49][50][51][52]. These CDT effects showed similarities with those exerted by some DNA damaging agents, such as ionizing radiation (IR) and etoposide, activating similar pathways [50][52][53]. In light of this observation, the apparent sequence homology between CdtB and the endonuclease/exonuclease/phosphatase family encouraged researchers to compare more precisely, among these proteins, CdtB with a well-known nuclease: deoxyribonuclease I (DNase I) [44][54]. As most of the DNase I residues essential for enzymatic activity are strikingly conserved in the different CdtB homologues, potential sites involved in the CdtB nuclease activity have been determined. The corresponding mutants failed to induce any distension, cell death or cell cycle arrest, showing that the CdtB catalytic activity is responsible for the observed cellular effects and that CdtB has a functional homology with DNase I [44][54]. Finally, the CdtB nuclease activity has been demonstrated in vitro by incubating a super-coiled plasmid DNA with the whole CDT holotoxin or with CdtB alone (see below). We reintroduce here the important concepts to study the DNA damage response pathway activation and relate them with the observations made after CDT treatment.
2.1. The CDT-Activated DNA Damage Response
In order to replicate correctly and to maintain the stability of their genetic information, eukaryotic cells display systems for controlling the genome integrity. Cells continuously undergo DNA damage generated by different sources (environment, metabolism, etc.). To survive in the presence of DNA lesions, the cell has developed mechanisms to detect and signal the damage. These interconnected pathways are referred to as the DNA damage response (DDR).
2.1.1. Introduction to the DDR
The DNA damage-related activation of checkpoints is divided into three stages: (i) the recognition of DNA damage by sensor proteins (MRN and Ku complexes, RPA), which rapidly activate specific phosphatidylinositol 3-kinase-related protein kinases (ATM, ATR, DNA-PK) [55]; (ii) the signal amplification by transducing proteins (CHK1, CHK2); which (iii) activates an appropriate cellular response by effectors proteins (p53, CDC25, etc.). This cell response initiates the cell cycle regulation, the activation of DNA repair pathways and, in some cases, cell death pathways [56].
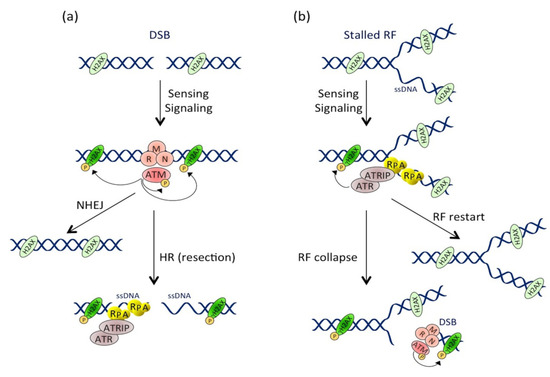
Two key signaling pathways are activated in response to DNA damage: the ATM-CHK2 and the ATR-CHK1 pathways. The ATM-CHK2 pathway is activated with the response to DSB-inducing agents. Following the generation of a DSB, the MRN complex, consisting of MRE11, RAD50 and NBS1, recruits the ATM kinase to the site of injury [57]. Once at the DSB site, ATM is activated by autophosphorylation and phosphorylates hundreds of substrates, including CHK2 and p53. Meanwhile, ATM phosphorylates the H2AX histone (then called γH2AX), several megabases around the DSB site [58], allowing signal amplification. The activated CHK2 phosphorylates various substrates, including p53 and the CDC25 phosphatases family. By contrast, the ATR-CHK1 pathway is activated by the accumulation of single-stranded DNA (ssDNA), particularly during the stalling of replication forks (RFs). Indeed, when replication is blocked by DNA lesions (SSB, DSB, inter-strand and intra-strand crosslinks, base modifications or adducts), DNA polymerase is uncoupled from the replicative helicase, which continues to unwind the DNA and, thus, generates ssDNA. ssDNA is recognized by the RPA protein complex, which protects and stabilizes it, and the accumulation of RPA-coated ssDNA at stalled RFs induces the recruitment of the ATR/ATRIP complex [59]. ATR then phosphorylates the CHK1 transducing protein [60]. ATR can, like ATM, phosphorylate H2AX [61], p53 and many cell cycle regulators (such as CDC25A, CDC25C and Wee1). In conclusion, whatever the activated pathway, the ATR-CHK1 or ATM-CHK2 activation will lead to major protein phosphorylation, involved in various cellular processes, including cell cycle regulation, DNA repair and programmed cell death [62]. However, it has to be underlined that many crosstalk exist between the ATM and ATR pathways (reviewed in [63]). Indeed, although CHK2 is the ATM primary target, ATM can also phosphorylate CHK1. In addition, processing of DSBs during the homologous recombination pathway (HR) generates stretches of ssDNA, leading to the ATR pathway activation [64]. Conversely, prolonged replicative stress can provoke RF collapse [65], resulting in DSB formation and ATM-CHK2 pathway activation (). In summary, according to the type of DNA injury, the ATM and ATR pathways can be specifically or sequentially activated, resulting in the cell cycle arrest, the activation of the DNA repair machinery and, potentially, in cell death.
2.1.2. CDT Activates the DNA Damage Response
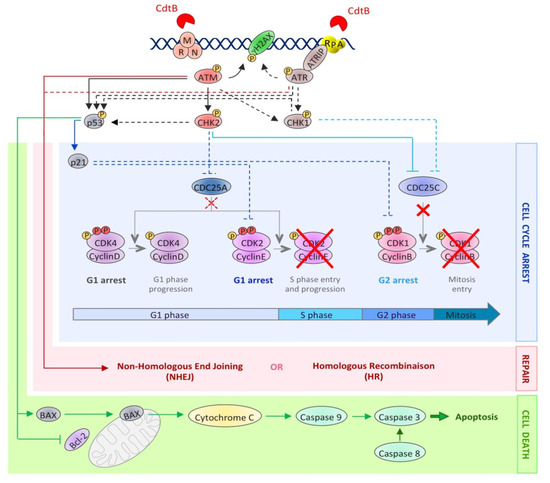
Many studies have compared the cellular effects of CdtB with DSB induced by IR. The first report deals with the cell cycle arrest induced in response to HdCDT or IR, in HL(human embryonic lung)-fibroblasts and HEp-2 cells [66], and suggested that CDT induces the activation of the DSB-related pathway. To better characterize the CdtB intracellular effects, the activation and the recruitment of different DDR proteins to damaged sites have been studied, demonstrating that CDT exposure recapitulates the different steps of DSB signaling (). First, the three subunits of the MRN complex have been shown to form nuclear foci following CDT exposure [67][68][69]. Different studies highlighted an activation of the ATM-dependent pathway, since the ATM protein level was increased and its phosphorylated form accumulated after CDT infection [11][53][70]. Besides, γH2AX foci are formed in response to CDT from various origins [53][67][68][71][72][73][74][75]. γH2AX is the most commonly used DSB biomarker, but can be observed in response to other stresses, such as replicative stress, hypoxia, chromatin remodeling, senescence or cell death [76]. Hence, to strengthen the fact that CDT induces DSB, another DSB marker, 53BP1, has been shown to form foci after CDT treatment [53][73][75]. Finally, ATM activation in response to CDT leads to CHK2 phosphorylation [11][53][70][72][73]. CHK2 phosphorylates different effectors, such as the cell cycle regulators of the phosphatase family, CDC25A and CDC25C. It is well known that ATM and CHK2 activate and stabilize p53, enhancing the transcription of the p21 gene involved in the cell cycle arrest at G1/S [77]. As expected, several studies showed that CDT induces p53 phosphorylation and stabilization, leading to the accumulation of p21 [38][73][78].
Figure 2. Activation and crosstalk between the ATM and ATR pathways. (a) Pathway activation at the double-stranded break (DSB). DSB formation induces the MRN-dependent ATM recruitment. ATM then phosphorylates numerous substrates, including itself and H2AX near the DSB site. DSB repair occurs through non-homologous end joining (NHEJ) or homologous recombination (HR) mechanisms. During HR, resection of the DSB extremities produces ssDNA stretches that are coated by RPA, leading to ATRIP-dependent ATR recruitment and activation. (b) Pathway activation during replicative stress. Replication fork (RF) stalling induces ssDNA formation that is rapidly coated by RPA. This structure is recognized by ATRIP, which drives the recruitment and activation of ATR. Besides phosphorylating its other substrates, ATR locally phosphorylates H2AX. RF can restart in case of transient stress or collapse after prolonged stresses. RF collapse induces DSB formation and ATM pathway activation, leading to a second and more important wave of H2AX phosphorylation.
Figure 3. The activation of the DNA damage response upon CDT exposure. This picture depicts the DDR molecular events induced after CDT intoxication. The dotted lines represent well-studied events occurring during the DDR, but not yet demonstrated in the context of CDT exposure. CdtB-induced DNA lesions are detected by sensors, such as the MRN complex and RPA, resulting in the recruitment and the activation of the PI3K related kinases, ATM and ATR. ATM and ATR then phosphorylate hundreds of substrates, including H2AX, CHK1, CHK2 and p53 (black arrows). This signaling cascade results in the regulation of cell cycle modulators (blue lines), through the inhibition of CDC25C phosphatase by CHK2 and, possibly, CHK1. Phosphorylated CDC25C is unable to activate the cyclin B/CDK1 complex (red crosses), essential for mitotic entry. Moreover, the p53-dependent accumulation of p21 blocks cells in G1 by inhibiting the CDK2/cyclin E complex. At the same time, the DSB repair mechanisms (NHEJ and HR) are activated by ATM and ATR (red arrows). However, if the level of DNA lesions is too severe, the process of cell death is initiated (green arrows). Apoptotic cell death can be induced by an intrinsic pathway involving p53 activation, leading to an increase in the BAX level, the sequestration and inactivation of Bcl-2, the mitochondrial release of cytochrome C and caspase 9 activation. Apoptosis can also be induced through the activation of the extrinsic pathway, involving caspase 8 activation. In both cases, this leads to caspase 3 activation and apoptotic cell death.
Few publications studied the activation of the ATR-CHK1 pathway in response to CDT. In 2006, Taieb et al. showed the phosphorylation of ATM, CHK2 and CHK1 proteins following a 24-h treatment with EcCDT. However, in this study, no information on the protein responsible for the CHK1 phosphorylation was given [72]. In a recent publication, the activation of the ATM-CHK2 and ATR-CHK1 pathways, in response to gamma-irradiation or CDT treatment, were compared [53]. Irradiated GM637 fibroblasts present a rapid and transient ATM-dependent CHK1 phosphorylation that precedes a second wave of CHK1 phosphorylation, mediated by ATR, which was not activated during the first 8 h post-irradiation. This delayed ATR-CHK1 pathway activation, as well as the late CHK2 phosphorylation are a consequence of the unrepaired DNA lesions that stall and ultimately collapse the RFs when cells progress through the S-phase. In contrast, following HdCDT treatment, the kinetics of ATR and CHK1 phosphorylation are totally different, while the ATM-CHK2 response is quite similar. Indeed, ATR is found phosphorylated at early time points after CDT exposure, with the same kinetics as CHK1 phosphorylation, which increases over time and is shown to be ATM-independent [53]. These data show that, in contrast to the IR-related DDR response, the ATR-CHK1 pathway is activated early and continuously in response to HdCDT.
2.2. Cellular Effects of the CDT-Induced DNA Damage
2.2.1. Cell Cycle Arrest
The cell-cycle progression is regulated by sequential activation and nuclear relocalization of CDK/Cyclin complexes [79]. At the beginning of the cell cycle, the activation of CDK4/Cyclin D and CDK2/Cyclin E complexes controls the G1-phase progression and entry into S-phase. The CDK2/Cyclin A complex then regulates the S-phase progression. The S-phase completion and G2 transition are coordinated by the activation of the CDK1/Cyclin A complex. Finally mitotic entry depends on the activation and nuclear relocalization of the CDK1/Cyclin B complex. CDK/Cyclin complexes are activated by CDC25-dependent dephosphorylation and can be inhibited by different regulators, like p16, p21, p27 and Wee1 [80].
In response to CDT treatment, CHK2 activation induces the sequestration of CDC25C phosphatase in the cytoplasm, making it unable to activate the CDK1/Cyclin B complex [81]. As a result, the CDK1/Cyclin B complex is hyperphosphorylated [11][52][72][82] and inactive [50], preventing cells from entering the G2-phase (). The implication of CDC25C in this process was demonstrated by overexpression of a recombinant CDC25C, which abrogates the CDT-induced G2/M cell cycle arrest [83]. Two other key factors of the cell cycle regulation are p53 and its p21 downstream target. Coherently, the G1/S checkpoint activation in response to CDT relies on the activation of p53 and p21 [84][85][86]. The G1/S checkpoint activation being largely dependent on p53, the variation in the p53 status among the cell types used in the experiments may explain the difference observed for their cell-cycle arrest; most of the p53 negative cells (or with a p53 inactive form) only arresting at G2/M and not at G1/S [68][74][86]. However, p21 induction can also be p53-independent, and a study indeed reported, in response to CDT, a p21 accumulation that was p53-independent [78]. In eukaryotic cells, p21 is known to inhibit many CDK/cyclin complexes, leading to the cell cycle regulation at different steps [87]. CDT intoxication may therefore lead to the cell cycle arrest at the G1/S transition via the CDK2/cyclin E inactivation.
2.2.2. Cell Death or Senescence
Cell cycle arrest is not the only cellular response induced by the DDR. Indeed, the ATM-CHK2 and ATR-CHK1 pathways also activate DNA repair and cell death pathways. Generally, CDT treatment leads to cell death by an apoptotic pathway. CDT-mediated apoptosis has been shown to follow the intrinsic mitochondrial pathway, involving the BAX/Bcl-2 protein [51][74][88], cytochrome C release [74] and caspases activation [74][88][89]. However, few cell lines were shown to use the extrinsic apoptotic pathway through the caspase-8 cleavage [88][89][90]. CDT intoxicated cells die either by apoptosis or necrosis, the latter being perhaps a consequence of abortive mitosis. Hence, some studies demonstrated that in spite of the DDR activation, CDT-treated cells can bypass the G2/M checkpoint, resulting in micronucleation and abortive mitosis [82][91]. Furthermore, endoreplication events have also been documented [49][82]. CDT has been shown to induce an apoptotic response (), following the cell cycle arrest, in a broad range of cell lines, with cell death being detectable between two and four days post-intoxication [9]. However, hematopoietic cells seem to activate a rapid apoptotic pathway during the first day of treatment to such an extent that the cell cycle arrest is not even observed [38][51][67][86][92]. When tested under the same conditions, non-hematopoietic cells presented a mild apoptotic response compared to hematopoietic cells, like monocytes and T-cells [93]. Hematopoietic cells therefore show the most dramatic apoptotic response and do not seem to activate the DDR in response to CDT, suggesting a specific cytotoxic mechanism that does not involve CDT-related DNA damage. Hence, such a DNA damage-independent cell death response may rely on the CdtB phosphatase activity [94].
Another possible cellular outcome of the CDT genotoxic effects is the induction of cellular senescence. CDT intoxicated cells express the hallmarks of cellular senescence (i.e., persistently activated DNA damage signaling, enhanced senescence-associated β-galactosidase activity and promyelocytic nuclear compartments expansion) [73]. This is especially important with regard to the senescence-associated secretory phenotype and inflammation [95], as the expression of proinflammatory mediators is observed after CDT infection [18][96]. Finally, this could lead to cytokine-induced bystander genotoxic effects, with a two-fold increase of reactive oxygen species (ROS) observed after a CDT chronic treatment [91].
to reflect the presence of both CdtA and CdtC. Different from all other CDTs, Salmonella enterica serovar Typhi CDT (SeCDT) has no CdtA and CdtC homologues. However, encoded closely to the active subunit cdtb, the Pertussis-like toxin A and B (pltA/pltB) have been shown to be essential for cellular intoxication.[10] PltA and PltB have a different structure from CdtA and CdtC, thus promoting CdtB activity in a different way. Both PltA and PltB have been found to bind directly to CdtB in vitro.[10] In addition, different from all other CDTs, Salmonella genotoxin is produced only upon bacterial internalization in infected cells, thus the SeCDT traffic may differ remarkably from the canonical ones.
6. CdtB
CdtB is considered the active subunit of the CDT holotoxin. Microinjection of CdtB into susceptible cells without CdtA or CdtC results in the G2/M cell cycle arrest and cytoplasmic distension characteristic of CDT toxins.[2] The structure of CdtB is well-conserved between different bacteria. The CdtB subunit is the most sequentially conserved between species.[4] The molecular weight of CdtB ranges from 28 kDa to 29 kDa, depending on the species.[1] As the active subunit, CdtB is termed the "A" subunit according to the AB toxin model.[1] This confusing nomenclature is due to the naming of the toxin's subunits before their individual functions were understood.
Activity
CdtB exhibits at least two enzymatic activities- DNase activity capable of introducing double-strand breaks in DNA, and a phosphatase activity that resembles phosphatidylinositol 3,4,5-triphosphatase.[2][8] Both activities can be demonstrated in vitro in the absence of the other two subunits.[11] The relative importance of each activity in vivo is unclear.[11] Mutations that reduce either activity also reduce the toxin's ability to induce G2/M phase arrest in at least some of the susceptible cell lines.[2][8]
Similarities to mammalian DNase I
CdtB is functionally homologous to mammalian DNase I and contains a conserved pentapeptide sequence found in all DNase I enzymes to date.[2] In addition, several residues critical to DNase I's ability to break the phosphodiester bonds in the DNA backbone are found in the CdtB structure. A 2002 paper studying the effect of point mutations on five of these residues found that four of the five mutations tested abolished both CdtB's ability to degrade DNA in cell-free extracts and to cause G2/M arrest upon microinjection. The fifth mutation moderately reduced CdtB's activity.[2]
7. CdtA and CdtC
CdtA and CdtC make up the B subunit of the CDT holotoxin responsible for targeting the CdtB against susceptible cells.[6] Neither subunit appears highly conserved, with sequence identities between different species often lower than 30%.[4] The molecular weight of CdtA ranges from 23 kDa to 30 kDa, whereas CdtC ranges from 19 kDa to 21 kDa depending on the species.[1]
Activity
CdtA and CdtC are both believed to bind to the surface of target cells. The exact mechanism of this binding is unclear, and may not be conserved between CDT toxins from different species.[1][11] Proposed targets of CdtA and CdtC binding have included cholesterol, N-linked glycans, and glycosphingolipids.[11] Current research has produced conflicting results on the actual importance of these proposed targets.[1][11] Both CdtA and CdtC contain lectin domains,[12] suggesting that the toxin may bind via carbohydrates on the target cell's surface, whereas other research has suggested that the targets are surface proteins.[1]
8. Notes
- ↑ 1.00 1.01 1.02 1.03 1.04 1.05 1.06 1.07 1.08 1.09 1.10 1.11 1.12 1.13 1.14 1.15 1.16 1.17 1.18 "Cytolethal distending toxin: a conserved bacterial genotoxin that blocks cell cycle progression, leading to apoptosis of a broad range of mammalian cell lineages". Microbiology 157 (Pt 7): 1851–75. July 2011. doi:10.1099/mic.0.049536-0. PMID 21565933. PMC 3167888. http://mic.sgmjournals.org/content/157/7/1851.short.
- ↑
- 2.0 2.1 2.2 2.3 2.4 2.5 2.6 Cherilyn A. Elwell, Lawrence A. Dreyfus (2000). "DNase I homologous residues in CdtB are critical for cytolethal distending toxin-mediated cell cycle arrest.". Molecular Microbiology 37 (4): 952–963. doi:10.1046/j.1365-2958.2000.02070.x.
- ↑ 3.0 3.1 3.2 Dreyfus, Lawrence, A. (2003), "Cyotlethal Distending Toxin", in D. Burns, Bacterial Protein Toxins, Washington, DC: ASM Press, pp. 257–270, https://books.google.com/books?hl=en&lr=&id=bQIsfnZTPOIC&oi=fnd&pg=PA257&dq=Cytolethal+distending+toxin&ots=4wf5ny_rt3&sig=htF6_cgjVw6-SEoNkGIT9FVltkM#v=onepage&q=Cytolethal%20distending%20toxin&f=false
- ↑ 4.0 4.1 4.2 4.3 "The biology of the cytolethal distending toxins". Toxins 3 (12): 172–90. March 2011. doi:10.3390/toxins3030172. PMID 22069704.
- ↑ 5.0 5.1 "Cloning and sequencing of the genes encoding Escherichia coli cytolethal distending toxin". Infection and Immunity 62 (1): 244–51. January 1994. PMID 8262635.
- ↑ 6.0 6.1 6.2 "CdtA, CdtB, and CdtC form a tripartite complex that is required for cytolethal distending toxin activity". Infection and Immunity 69 (7): 4358–65. July 2001. doi:10.1128/IAI.69.7.4358-4365.2001. PMID 11401974.
- ↑ 7.0 7.1 Cortes-Bratti, Teresa Frisan, Monica Thelestam. (2001). "The Cytolethal Distending Toxins Induce DNA Damage and Cell Cycle Arrest.". Toxicon 39 (11): 1729–1736. doi:10.1016/S0041-0101(01)00159-3.
- ↑ 8.0 8.1 8.2 8.3 8.4 Bruce J. Shenker, Mensur Dlakic, Lisa P. Walker, Dave Besack, Eileen Jaffe, Ed LaBelle, Kathleen Boesze-Battaglia. (2007). "A Novel Mode of Action for a Microbial-Derived Immunotoxin: The Cytolethal Distending Toxin Subunit B Exhibits Phosphatidylinositol 3,4,5-Triphosphate Phosphatase Activity". The Journal of Immunology 178 (8): 5099–5108. doi:10.4049/jimmunol.178.8.5099. PMID 17404292.
- ↑ "Bacterial intoxication evokes cellular senescence with persistent DNA damage and cytokine signalling". Journal of Cellular and Molecular Medicine 14 (1–2): 357–67. January 2010. doi:10.1111/j.1582-4934.2009.00862.x. PMID 19650831.
- ↑
- 10.0
-
- 10.1
-
- "Delivery of a Salmonella Typhi exotoxin from a host intracellular compartment".
- Cell Host & Microbe
-
- 3
- (1): 30–8. January 2008. doi:10.1016/j.chom.2007.11.001. PMID 18191792.
- ↑ 11.0 11.1 11.2 11.3 11.4 "Cytolethal distending toxin family members are differentially affected by alterations in host glycans and membrane cholesterol". The Journal of Biological Chemistry 285 (24): 18199–207. June 2010. doi:10.1074/jbc.m110.112912. PMID 20385557.
- ↑ "Assembly and function of a bacterial genotoxin". Nature 429 (6990): 429–33. May 2004. doi:10.1038/nature02532. PMID 15164065.