The cytolethal distending toxin (CDT) is produced by many pathogenic Gram-negative bacteria and is considered as a virulence factor. In human cells, CDT exposure leads to a unique cytotoxicity associated with a characteristic cell distension and induces a cell cycle arrest dependent on the DNA damage response (DDR) triggered by DNA double-strand breaks (DSBs). CDT has thus been classified as a cyclomodulin and a genotoxin.
- Gram-negative bacteria
- cytolethal distending toxin
- DNA damage response
- double-strand breaks
- cell cycle checkpoints
- replicative stress
1. History
The first recorded observation of a cytolethal-distending toxin was in 1987 in a pathogenic strain in E. coli isolated from a young patient.[3] Later that year, scientists W.M. Johnson and H. Lior published the journal article "Production of Shiga toxin and a cytolethal distending toxin (CLDT) by serogroups of Shigella spp." in Microbiology Letters.[1] The discovery of other bacteria producing CDT toxins continues to this day. In 1994 Scott and Kaper cloned and sequenced a cdt operon from another E. coli strain, publishing in Infection and Immunity.[1][5] The three genes discovered were denoted cdtA, cdtB, and cdtC.[5] In 1997, the first paper of many to show G2/M cell cycle arrest caused by a cytolethal distending toxin was published in Molecular Microbiology.[1] The study focused on another E. coli strain. This paper was followed by a 1999 publication in Infectious Immunity, which demonstrated that H. ducreyi CDT causes cell death via apoptosis. This finding was also confirmed for other cytolethal distending toxins in subsequent studies. The discovery of the homology of cdtB to mammalian DNase I and the current AB model for the toxin were published in early 2000. [2][6] Further research and the publication of crystal structures for the CDT toxins from two different species continues to support this model.[1]
2. Sources
All known cytolethal distending toxins are produced by gram-negative bacteria in the Gammaproteobacteria and Campylobacterota. In several cases, the bacteria producing CDT are human pathogens. Medically important CDT producers include:[1]
- Haemophilus ducreyi (chancroids)
- Aggregatibacter actinomycetemcomitans (periodontitis)
- Escherichia coli (various diseases)
- Shigella dysenteriae (dysentery)
- Salmonella enterica serotype Typhi (typhoid fever)
- Campylobacter upsaliensis (enterocolitis)
- Campylobacter jejuni (enterocolitis)
CDT-producing bacteria are often associated with mucosal linings, such as those in the stomach and intestines, and with persistent infections. The toxins are either secreted freely or associated with the membrane of the producing bacteria.[1]
3. Nomenclature
Individual cytolethal distending toxins are named for the bacterial species that they are isolated from. As of 2011, most scientists have adopted the practice of placing the first letter of both the genus and species in front of the toxin name to reflect its source (i.e., the CDT from Haemaphilus ducreyi is referred to as HdCDT).[1][7] If several subspecies produce different toxins, as in the case of E. coli, Roman numerals may be added after the second letter.[7] Both complete toxins and individual subunits are labeled using this convention. In response to the continued discovery of additional cytolethal distending toxins, a 2011 review has proposed that the toxin names be expanded to include the first three letters of the species (i.e., HducCDT for Haemaphilus ducreyi CDT).[1]
4. Cellular Effects
CDT toxins are genotoxins capable of directly damaging DNA in target cells. They are the only AB-type toxins discovered that display DNase activity, allowing them to introduce breaks into the target cell's DNA.[1][4] In many cell lines including human fibroblasts, epithelial cells, endothelial cells, and keratinocytes, CDTs cause G2/M cell cycle arrest, cytoplasmic distension, and eventual cell death via apoptosis.[1][3][8] Most publications attribute the G2/M cycle arrest to the buildup of irreversible DNA damage from the toxin's DNase activity as the trigger for the G2/M cell cycle arrest, but other research suggests that this model is incomplete.[8] The cytoplasmic distension is a direct result of the G2/M cell cycle arrest. The cell enlarges in preparation for mitosis, but cannot divide to restore its normal size. Aside from classical apoptosis, signs of cellular senescence has also been observed in normal and cancer cell lines (fibroblasts, HeLa and U2-OS) after CDT intoxication[9] In lymphocytes, cell death occurs quickly and is not preceded by significant cytoplasmic distension.[8] The ability of these toxins to effect lymphocytes differently may be advantageous to the bacteria that utilize these toxins, but the mechanism behind this phenomenon is not yet well understood.
5. Toxin Structure
The active, assembled toxin is a tripartite structure with three distinct subunits- CdtA, CdtB, and CdtC. In terms of function, it is an AB toxin. In this context, the CdtB subunit is actually the catalytically active "A" subunit, and the CdtA and CdtC together form the binding "B" subunit, which helps the toxin bind and enter target cells.[6] Some literature refers to the toxin structure as AB
1. Introduction
1.1. CDT-Related Pathogenicity
1.2. CDT is a Tripartite A-B Exotoxin
2 to reflect the presence of both CdtA and CdtC. Different from all other CDTs, Salmonella enterica serovar Typhi CDT (SeCDT) has no CdtA and CdtC homologues. However, encoded closely to the active subunit cdtb, the Pertussis-like toxin A and B (pltA/pltB) have been shown to be essential for cellular intoxication.[10] PltA and PltB have a different structure from CdtA and CdtC, thus promoting CdtB activity in a different way. Both PltA and PltB have been found to bind directly to CdtB in vitro.[10] In addition, different from all other CDTs, Salmonella genotoxin is produced only upon bacterial internalization in infected cells, thus the SeCDT traffic may differ remarkably from the canonical ones.
6. CdtB
CdtB is considered the active subunit of the CDT holotoxin. Microinjection of CdtB into susceptible cells without CdtA or CdtC results in the G2/M cell cycle arrest and cytoplasmic distension characteristic of CDT toxins.[2] The structure of CdtB is well-conserved between different bacteria. The CdtB subunit is the most sequentially conserved between species.[4] The molecular weight of CdtB ranges from 28 kDa to 29 kDa, depending on the species.[1] As the active subunit, CdtB is termed the "A" subunit according to the AB toxin model.[1] This confusing nomenclature is due to the naming of the toxin's subunits before their individual functions were understood.
Activity
CdtB exhibits at least two enzymatic activities- DNase activity capable of introducing double-strand breaks in DNA, and a phosphatase activity that resembles phosphatidylinositol 3,4,5-triphosphatase.[2][8] Both activities can be demonstrated in vitro in the absence of the other two subunits.[11] The relative importance of each activity in vivo is unclear.[11] Mutations that reduce either activity also reduce the toxin's ability to induce G2/M phase arrest in at least some of the susceptible cell lines.[2][8]
Similarities to mammalian DNase I
CdtB is functionally homologous to mammalian DNase I and contains a conserved pentapeptide sequence found in all DNase I enzymes to date.[2] In addition, several residues critical to DNase I's ability to break the phosphodiester bonds in the DNA backbone are found in the CdtB structure. A 2002 paper studying the effect of point mutations on five of these residues found that four of the five mutations tested abolished both CdtB's ability to degrade DNA in cell-free extracts and to cause G2/M arrest upon microinjection. The fifth mutation moderately reduced CdtB's activity.[2]
7. CdtA and CdtC
CdtA and CdtC make up the B subunit of the CDT holotoxin responsible for targeting the CdtB against susceptible cells.[6] Neither subunit appears highly conserved, with sequence identities between different species often lower than 30%.[4] The molecular weight of CdtA ranges from 23 kDa to 30 kDa, whereas CdtC ranges from 19 kDa to 21 kDa depending on the species.[1]
Activity
CdtA and CdtC are both believed to bind to the surface of target cells. The exact mechanism of this binding is unclear, and may not be conserved between CDT toxins from different species.[1][11] Proposed targets of CdtA and CdtC binding have included cholesterol, N-linked glycans, and glycosphingolipids.[11] Current research has produced conflicting results on the actual importance of these proposed targets.[1][11] Both CdtA and CdtC contain lectin domains,[12] suggesting that the toxin may bind via carbohydrates on the target cell's surface, whereas other research has suggested that the targets are surface proteins.[1]
8. Notes
- ↑ 1.00 1.01 1.02 1.03 1.04 1.05 1.06 1.07 1.08 1.09 1.10 1.11 1.12 1.13 1.14 1.15 1.16 1.17 1.18 "Cytolethal distending toxin: a conserved bacterial genotoxin that blocks cell cycle progression, leading to apoptosis of a broad range of mammalian cell lineages". Microbiology 157 (Pt 7): 1851–75. July 2011. doi:10.1099/mic.0.049536-0. PMID 21565933. PMC 3167888. http://mic.sgmjournals.org/content/157/7/1851.short.
- ↑ 2.0 2.1 2.2 2.3 2.4 2.5 2.6 Cherilyn A. Elwell, Lawrence A. Dreyfus (2000). "DNase I homologous residues in CdtB are critical for cytolethal distending toxin-mediated cell cycle arrest.". Molecular Microbiology 37 (4): 952–963. doi:10.1046/j.1365-2958.2000.02070.x.
- ↑ 3.0 3.1 3.2 Dreyfus, Lawrence, A. (2003), "Cyotlethal Distending Toxin", in D. Burns, Bacterial Protein Toxins, Washington, DC: ASM Press, pp. 257–270, https://books.google.com/books?hl=en&lr=&id=bQIsfnZTPOIC&oi=fnd&pg=PA257&dq=Cytolethal+distending+toxin&ots=4wf5ny_rt3&sig=htF6_cgjVw6-SEoNkGIT9FVltkM#v=onepage&q=Cytolethal%20distending%20toxin&f=false
- ↑ 4.0 4.1 4.2 4.3 "The biology of the cytolethal distending toxins". Toxins 3 (12): 172–90. March 2011. doi:10.3390/toxins3030172. PMID 22069704.
- ↑ 5.0 5.1 "Cloning and sequencing of the genes encoding Escherichia coli cytolethal distending toxin". Infection and Immunity 62 (1): 244–51. January 1994. PMID 8262635.
- ↑ 6.0 6.1 6.2 "CdtA, CdtB, and CdtC form a tripartite complex that is required for cytolethal distending toxin activity". Infection and Immunity 69 (7): 4358–65. July 2001. doi:10.1128/IAI.69.7.4358-4365.2001. PMID 11401974.
- ↑ 7.0 7.1 Cortes-Bratti, Teresa Frisan, Monica Thelestam. (2001). "The Cytolethal Distending Toxins Induce DNA Damage and Cell Cycle Arrest.". Toxicon 39 (11): 1729–1736. doi:10.1016/S0041-0101(01)00159-3.
- 11.0 11.1 11.2 11.3 11.4 "Cytolethal distending toxin family members are differentially affected by alterations in host glycans and membrane cholesterol". The Journal of Biological Chemistry 285 (24): 18199–207. June 2010. doi:10.1074/jbc.m110.112912. PMID 20385557.
- ↑ "Assembly and function of a bacterial genotoxin". Nature 429 (6990): 429–33. May 2004. doi:10.1038/nature02532. PMID 15164065.
 |
- ↑
- 8.0
- 8.1
- 8.2
- 8.3 8.4 Bruce J. Shenker, Mensur Dlakic, Lisa P. Walker, Dave Besack, Eileen Jaffe, Ed LaBelle, Kathleen Boesze-Battaglia. (2007). "A Novel Mode of Action for a Microbial-Derived Immunotoxin: The Cytolethal Distending Toxin Subunit B Exhibits Phosphatidylinositol 3,4,5-Triphosphate Phosphatase Activity". The Journal of Immunology 178 (8): 5099–5108. doi:10.4049/jimmunol.178.8.5099. PMID 17404292.
- ↑ "Bacterial intoxication evokes cellular senescence with persistent DNA damage and cytokine signalling". Journal of Cellular and Molecular Medicine 14 (1–2): 357–67. January 2010. doi:10.1111/j.1582-4934.2009.00862.x. PMID 19650831.
- ↑ 10.0 10.1 "Delivery of a Salmonella Typhi exotoxin from a host intracellular compartment". Cell Host & Microbe 3 (1): 30–8. January 2008. doi:10.1016/j.chom.2007.11.001. PMID 18191792.
- ↑
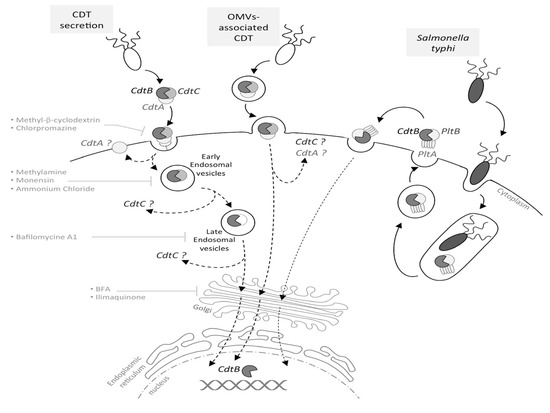
1.3. From CDT Host Cell Binding to CdtB Nuclear Localization
2. DNA Damage-Related Cellular Outcomes of CDT Intoxication
Since the discovery of CDT, the cellular response to CDT intoxication has been better and better characterized. CDT induces a cell cycle arrest (at the G2/M transition and, depending on the cell type, at the G1/S transition), accompanied by a cellular distention and, eventually, cell death [49][50][51][52]. These CDT effects showed similarities with those exerted by some DNA damaging agents, such as ionizing radiation (IR) and etoposide, activating similar pathways [50][52][53]. In light of this observation, the apparent sequence homology between CdtB and the endonuclease/exonuclease/phosphatase family encouraged researchers to compare more precisely, among these proteins, CdtB with a well-known nuclease: deoxyribonuclease I (DNase I) [44][54]. As most of the DNase I residues essential for enzymatic activity are strikingly conserved in the different CdtB homologues, potential sites involved in the CdtB nuclease activity have been determined. The corresponding mutants failed to induce any distension, cell death or cell cycle arrest, showing that the CdtB catalytic activity is responsible for the observed cellular effects and that CdtB has a functional homology with DNase I [44][54]. Finally, the CdtB nuclease activity has been demonstrated in vitro by incubating a super-coiled plasmid DNA with the whole CDT holotoxin or with CdtB alone (see below). We reintroduce here the important concepts to study the DNA damage response pathway activation and relate them with the observations made after CDT treatment.2.1. The CDT-Activated DNA Damage Response
2.1.1. Introduction to the DDR
2.1.2. CDT Activates the DNA Damage Response
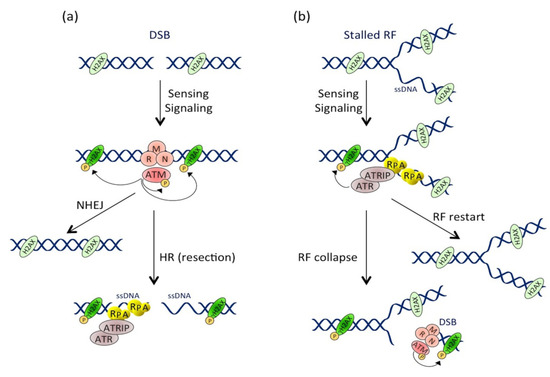
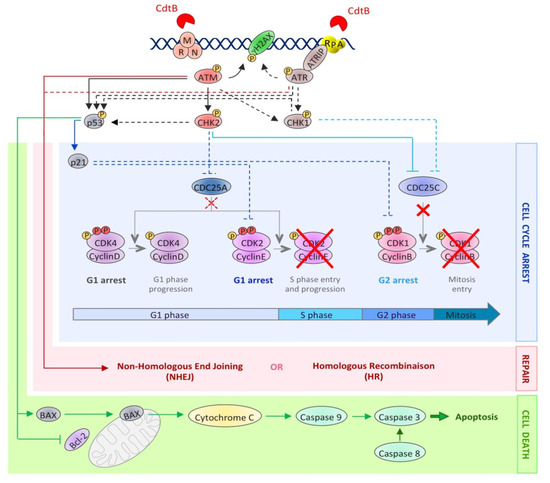
2.2. Cellular Effects of the CDT-Induced DNA Damage
2.2.1. Cell Cycle Arrest
2.2.2. Cell Death or Senescence
References
- Johnson, W.M.; Lior, H. Response of chinese hamster ovary cells to a cytolethal distending toxin (CDT) of Escherichia coli and possible misinterpretation as heat-labile (LT) enterotoxin. FEMS Microbiol. Lett. 1987, 43, 19–23.
- Johnson, W.M.; Lior, H. A new heat-labile Cytolethal Distending Toxin (CLDT) produced by campylobacter spp. Microbiol. Pathol. 1988, 4, 115–126.
- Cope, L.D.; Lumbley, S.; Latimer, J.L.; Klesney-Tait, J.; Stevens, M.K.; Johnson, L.S.; Purven, M.; Munson, R.S.; Lagergard, T.; Radolf, J.D.; et al. A diffusible cytotoxin of haemophilus ducreyi. Proc. Natl. Acad. Sci. 1997, 94, 4056–4061.
- Sugai, M.; Kawamoto, T.; Pérès, S.Y.; Ueno, Y.; Komatsuzawa, H.; Fujiwara, T.; Kurihara, H.; Suginaka, H.; Oswald, E. The cell cycle-specific growth-inhibitory factor produced by actinobacillus actinomycetemcomitans is a cytolethal distending toxin. Infect. Immun. 1998, 66, 5008–5019.
- Shen, Z.; Feng, Y.; Rogers, A.B.; Rickman, B.; Whary, M.T.; Xu, S.; Clapp, K.M.; Boutin, S.R.; Fox, J.G. Cytolethal distending toxin promotes helicobacter cinaedi-associated typhlocolitis in interleukin-10-deficient mice. Infect. Immun. 2009, 77, 2508–2516.
- Haghjoo, E.; Galán, J.E. Salmonella typhi encodes a functional cytolethal distending toxin that is delivered into host cells by a bacterial-internalization pathway. Proc. Natl. Acad. Sci. 2004, 101, 4614–4619.
- Stanley, J.; Linton, D.; Burnens, A.P.; Dewhirst, F.E.; On, S.L.W.; Porter, A.; Owen, R.J.; Costas, M. Helicobacter pullorurn sp. nov.-genotype and phenotype of a new species isolated from poultry and from human patients with gastroenteritis. Microbiology 1994, 140, 3441–3449.
- Chien, C.C.; Taylor, N.S.; Ge, Z.; Schauer, D.B.; Young, V.B.; Fox, J.G. Identification of cdtB homologues and cytolethal distending toxin activity in enterohepatic Helicobacter spp. J. Med. Microbiol. 2000, 49, 525–534.
- Jinadasa, R.N.; Bloom, S.E.; Weiss, R.S.; Duhamel, G.E. Cytolethal distending toxin: A conserved bacterial genotoxin that blocks cell cycle progression , leading to apoptosis of a broad range of mammalian cell lineages. Microbiology 2011, 157, 1851–1875.
- Nipic, D.; Podlesek, Z.; Budic, M.; Crnigoj, M.; Zgur-Bertok, D. Escherichia coli uropathogenic-specific protein, usp, is a bacteriocin-like genotoxin. J. Infect. Dis. 2013, 208, 1545–1552.
- Nougayrède, J.; Homburg, S.; Taieb, F.; Boury, M.; Brzuszkiewicz, E.; Gottschalk, G.; Buchrieser, C.; Hacker, J.; Dobrindt, U.; Oswald, E. Escherichia coli induces dna double-strand breaks in eukaryotic cells. Science 2006, 313, 848–851.
- Yamamoto, S.; Nakano, M.; Terai, A.; Yuri, K.; Nakata, K.; Nair, G.B.; Kurazono, H.; Ogawa, O. The presence of the virulence island containing the usp gene in uropathogenic Escherichia coli is associated with urinary tract infection in an experimental mouse model. J. Urol. 2001, 165, 1347–1351.
- Arthur, J.C.; Perez-chanona, E.; Mühlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012, 338, 120–123.
- Finlay, B.B.; Falkow, S. Common themes in microbial pathogenicity revisited. Microbiol. Mol. Biol. Rev. 1997, 61, 136–169.
- Ge, Z.; Schauer, D.B.; Fox, J.G. In vivo virulence properties of bacterial Cytolethal Distending Toxin. Cell. Microbiol. 2008, 10, 1599–1607.
- Jain, D.; Prasad, K.N.; Sinha, S.; Husain, N. Differences in virulence attributes between cytolethal distending toxin positive and negative campylobacter jejuni strains. J. Med. Microbiol. 2008, 57, 267–272.
- Pokkunuri, V.; Pimentel, M.; Morales, W.; Jee, S.R.; Alpern, J.; Weitsman, S.; Marsh, Z.; Low, K.; Hwang, L.; Khoshini, R.; et al. Hole of cytolethal distending toxin in altered stool form and bowel phenotypes in a rat model of post-infectious irritable bowel syndrome. J. Neurogastroenterol. Motil. 2012, 18, 434–442.
- Ge, Z.; Rogers, A.B.; Feng, Y.; Lee, A.; Xu, S.; Taylor, N.S.; Fox, J.G. Bacterial cytolethal distending toxin promotes the development of dysplasia in a model of microbially induced hepatocarcinogenesis. Cell. Microbiol. 2007, 9, 2070–2080.
- Song, J.; Gao, X.; Galán, J.E. Structure and function of the salmonella typhi chimaeric A2B5 typhoid toxin. Nature 2013, 499, 350–354.
- Scott, D.A.; Kaper, J.B. Cloning and sequencing of the genes encoding Escherichia coli cytolethal distending toxin. Infect. Immun. 1994, 62, 244–251.
- Pickett, C.L.; Cottle, D.L.; Pesci, E.C.; Bikah, G. Cloning, sequencing, and expression of the Escherichia coli cytolethal distending toxin genes. Infect. Immun. 1994, 62, 1046–1051.
- Nesić, D.; Hsu, Y.; Stebbins, C.E. Assembly and function of a bacterial genotoxin. Nature 2004, 429, 429–433.
- Yamada, T.; Komoto, J.; Saiki, K.; Konishi, K.; Takusagawa, F. Variation of loop sequence alters stability of cytolethal distending toxin (CDT): Crystal structure of CDT from actinobacillus actinomycetemcomitans. Protein Sci. 2006, 15, 362–372.
- Spanò, S.; Ugalde, J.E.; Galán, J.E. Delivery of a salmonella typhi exotoxin from a host intracellular compartment. Cell Host Microbe 2008, 3, 30–38.
- Kaslow, H.R.; Burns, D.L. Pertussis toxin and target eukaryotic cells: Binding, entry, and activation. FASEB J. 1992, 6, 2685–2690.
- Ueno, Y.; Ohara, M.; Kawamoto, T.; Fujiwara, T.; Komatsuzawa, H.; Oswald, E.; Sugai, M. Biogenesis of the actinobacillus actinomycetemcomitans cytolethal distending toxin holotoxin. Infect. Immun. 2006, 74, 3480–3487.
- Zijnge, V.; Kieselbach, T.; Oscarsson, J. Proteomics of protein secretion by aggregatibacter actinomycetemcomitans. PLoS One 2012, 7, e41662.
- Berlanda Scorza, F.; Doro, F.; Rodríguez-Ortega, M.J.; Stella, M.; Liberatori, S.; Taddei, A.R.; Serino, L.; Gomes Moriel, D.; Nesta, B.; Fontana, M.R.; et al. Proteomics characterization of outer membrane vesicles from the extraintestinal pathogenic Escherichia coli deltatolR IHE3034 mutant. Mol. Cell. Proteomics 2008, 7, 473–485.
- Lindmark, B.; Rompikuntal, P.K.; Vaitkevicius, K.; Song, T.; Mizunoe, Y.; Uhlin, B.E.; Guerry, P.; Wai, S.N. Outer membrane vesicle-mediated release of cytolethal distending toxin (CDT) from campylobacter jejuni. BMC Microbiol. 2009, 9, 220.
- Rompikuntal, P.K.; Thay, B.; Khan, M.K.; Alanko, J.; Penttinen, A.-M.; Asikainen, S.; Wai, S.N.; Oscarsson, J. Perinuclear localization of internalized outer membrane vesicles carrying active cytolethal distending toxin from aggregatibacter actinomycetemcomitans. Infect. Immun. 2012, 80, 31–42.
- Gargi, A.; Reno, M.; Blanke, S.R. Bacterial toxin modulation of the eukaryotic cell cycle: Are all cytolethal distending toxins created equally? Front. Cell. Infect. Microbiol. 2012, 2, 124.
- Hu, X.; Nesic, D.; Stebbins, C.E. Comparative structure-function analysis of cytolethal distending toxins. Proteins 2006, 62, 421–434.
- Lee, R.B.; Hassane, D.C.; Cottle, D.L.; Pickett, C.L. Interactions of campylobacter jejuni cytolethal distending toxin subunits CdtA and CdtC with HeLa cells. Infect. Immun. 2003, 71, 4883–4890.
- Mcsweeney, L.A.; Dreyfus, L.A. Carbohydrate-binding specificity of the Escherichia coli cytolethal distending toxin CdtA-II and CdtC-II subunits. Infect. Immun. 2005, 73, 2051–2060.
- Eshraghi, A.; Maldonado-arocho, F.J.; Gargi, A.; Cardwell, M.M.; Prouty, M.G.; Blanke, S.R.; Bradley, K.A. Cytolethal distending toxin family members are differentially affected by alterations in host glycans and membrane cholesterol. Microbiology 2010, 285, 18199–18207.
- Carette, J.E.; Guimaraes, C.P.; Varadarajan, M.; Park, A.S.; Wuethrich, I.; Godarova, A.; Kotecki, M.; Cochran, B.H.; Spooner, E.; Ploegh, H.L.; et al. Haploid genetic screens in human cells identify host factors used by pathogens. Science 2009, 326, 1231–1235.
- Carette, J.E.; Guimaraes, C.P.; Wuethrich, I.; Blomen, V.A.; Sun, C.; Bell, G.; Yuan, B.; Muellner, M.K.; Nijman, M.; Ploegh, H.L.; et al. Global gene disruption in human cells to assign genes to phenotypes. Nat. Biotechnol. 2011, 29, 542–546.
- Cortes-Bratti, X.; Chaves-Olarte, E.; Lagergård, T.; Thelestam, M. Cellular Internalization of cytolethal distending toxin from haemophilus ducreyi. Infect. Immun. 2000, 68, 6903–6911.
- Guerra, L.; Teter, K.; Lilley, B.N.; Stenerlöw, B.; Holmes, R.K.; Ploegh, H.L.; Sandvig, K.; Thelestam, M.; Frisan, T. Cellular internalization of cytolethal distending toxin: A new end to a known pathway. Cell. Microbiol. 2005, 7, 921–934.
- Boesze-Battaglia, K.; Besack, D.; McKay, T.; Zekavat, A.; Otis, L.; Jordan-Sciutto, K.; Shenker, B.J. Cholesterol-rich membrane microdomains mediate cell cycle arrest induced by actinobacillus actinomycetemcomitans cytolethal-distending toxin. Cell. Microbiol. 2006, 8, 823–836.
- Boesze-Battaglia, K.; Brown, A.; Walker, L.; Besack, D.; Zekavat, A.; Wrenn, S.; Krummenacher, C.; Shenker, B.J. cytolethal distending toxin-induced cell cycle arrest of lymphocytes is dependent upon recognition and binding to cholesterol. J. Biol. Chem. 2009, 284, 10650–10658.
- Lin, C.D.; Lai, C.K.; Lin, Y.H.; Hsieh, J.T.; Sing, Y.T.; Chang, Y.C.; Chen, K.C.; Wang, W.C.; Su, H.L.; Lai, C.H. Cholesterol depletion reduces entry of Campylobacter jejuni cytolethal distending toxin and attenuates intoxication of host cells. Infect. Immun. 2011, 79, 3563–3575.
- Damek-Poprawa, M.; Jang, J.Y.; Volgina, A.; Korostoff, J.; DiRienzo, J.M. Localization of Aggregatibacter actinomycetemcomitans cytolethal distending toxin subunits during intoxication of live cells. Infect. Immun. 2012, 80, 2761–2770.
- Lara-Tejero, M.; Galàn, J.E. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-Like protein. Science 2000, 290, 354–357.
- Nishikubo, S.; Ohara, M.; Ueno, Y.; Ikura, M.; Kurihara, H.; Komatsuzawa, H.; Oswald, E.; Sugai, M. Ann-terminal segment of the active component of the bacterial genotoxin cytolethal distending toxin B (CDTB) directs CDTB into the nucleus. J. Biol. Chem. 2003, 278, 50671–50681.
- Guerra, L.; Nemec, K.N.; Massey, S.; Tatulian, S.A.; Thelestam, M.; Frisan, T.; Teter, K. A novel mode of translocation for cytolethal distending toxin. Biochim. Biophys. Acta 2009, 1793, 489–495.
- Gargi, A.; Tamilselvam, B.; Powers, B.; Prouty, M.G.; Lincecum, T.; Eshraghi, A.; Maldonado-Arocho, F.J.; Wilson, B.A.; Bradley, K.A.; Blanke, S.R. Cellular interactions of the cytolethal distending toxins from Escherichia coli and haemophilus ducreyi. J. Biol. Chem. 2013, 288, 7492–7505.
- McSweeney, L.A.; Dreyfus, L.A. A Nuclear localization of the Escherichia coli cytolethal Distending toxin CdtB subunit. Cell. Microbiol. 2004, 6, 447–458.
- Pérès, S.Y.; Marchès, O.; Daigle, F.; Nougayrède, J.P.; Herault, F.; Tasca, C.; de Rycke, J.; Oswald, E. A new cytolethal distending toxin (CDT) from Escherichia coli producing CNF2 blocks HeLa cell division in G2/M phase. Mol. Microbiol. 1997, 24, 1095–1107.
- Comayras, C.; Tasca, C.; Pe, S.Y.; Oswald, E.; Rycke, J.D.E. Escherichia coli cytolethal distending toxin blocks the HeLa cell cycle at the G2/M transition by preventing cdc2 protein kinase dephosphorylation and activation. Escherichia coli cytolethal distending toxin blocks the HeLa cell cycle at the G 2/M Tr. Infect. Immun. 1997, 65, 5088–5095.
- Ohguchi, M.; Ishisaki, A.; Okahashi, N.; Koide, M.; Koseki, T.; Yamato, K.; Noguchi, T.; Nishihara, T. Actinobacillus actinomycetemcomitans toxin induces both cell cycle arrest in the G2/M phase and apoptosis. Infect. Immun. 1998, 66, 5980–5987.
- Sert, V.; Cans, C.; Tasca, C.; Oswald, E.; Ducommun, B.; de Rycke, J. The bacterial cytolethal distending toxin (CDT) triggers a G2 cell cycle checkpoint in mammalian cells without preliminary induction of DNA strand breaks. Oncogene 1999, 18, 6296–6304.
- Fahrer, J.; Huelsenbeck, J.; Jaurich, H.; Dörsam, B.; Frisan, T.; Eich, M.; Roos, W.P.; Kaina, B.; Fritz, G. Cytolethal distending toxin (CDT) is a radiomimetic agent and induces persistent levels of DNA double-strand breaks in human fibroblasts. DNA Repair (Amst) 2014, 18, 31–43.
- Elwell, C.A.; Dreyfus, L.A. DNase I homologous residues in CdtB are critical for cytolethal distending toxin-mediated cell cycle arrest. Mol. Microbiol. 2000, 37, 952–963.
- Niida, H.; Nakanishi, M. DNA damage checkpoints in mammals. Mutagenesis 2006, 21, 3–9.
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078.
- Lee, J.-H.; Paull, T.T. ATM activation by DNA double-Strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005, 308, 551–554.
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999, 146, 905–916.
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548.
- Zhao, H.; Piwnica-Worms, H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell. Biol. 2001, 21, 4129–4139.
- Ward, I.M.; Chen, J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 2001, 276, 47759–47762.
- Matsuoka, S.; Ballif, B.A; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166.
- Gobbini, E.; Cesena, D.; Galbiati, A.; Lockhart, A.; Longhese, M.P. Interplays between ATM/Tel1 and ATR/Mec1 in sensing and signaling DNA double-strand breaks. DNA Repair (Amst) 2013, 12, 791–799.
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.C.M.; Lukas, J.; Jackson, S.P. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 37–45.
- Feng, W.; di Rienzi, S.C.; Raghuraman, M.K.; Brewer, B.J. Replication stress-induced chromosome breakage is correlated with replication fork progression and is preceded by single-stranded DNA formation. G3 Genes Genomes Genet. 2011, 1, 327–335.
- Cortes-Bratti, X.; Karlsson, C.; Lagergård, T.; Thelestam, M.; Frisan, T. The haemophilus ducreyi cytolethal distending Toxin induces cell cycle arrest and apoptosis via the DNA damage checkpoint pathways. J. Biol. Chem. 2001, 276, 5296–5302.
- Li, L.; Sharipo, A.; Chaves-Olarte, E.; Masucci, M.G.; Levitsky, V.; Thelestam, M.; Frisan, T. The haemophilus ducreyi cytolethal distending toxin activates sensors of DNA damage and repair complexes in proliferating and non-proliferating cells. Cell. Microbiol. 2002, 4, 87–99.
- Hassane, D.C.; Lee, R.B.; Pickett, C.L. Campylobacter jejuni cytolethal distending toxin promotes DNA repair responses in normal human cells. Infect. Immun. 2003, 71, 541–545.
- Guerra, L.; Albihn, A.; Tronnersjö, S.; Yan, Q.; Guidi, R.; Stenerlöw, B.; Sterzenbach, T.; Josenhans, C.; Fox, J.G.; Schauer, D.B.; et al. Myc is required for activation of the ATM-dependent checkpoints in response to DNA damage. PLoS One 2010, 5, e8924.
- Alaoui-El-Azher, M.; Mans, J.J.; Baker, H.V; Chen, C.; Progulske-Fox, A.; Lamont, R.J.; Handfield, M. Role of the ATM-checkpoint kinase 2 pathway in CDT-mediated apoptosis of gingival epithelial cells. PLoS One 2010, 5, e11714.
- Bielaszewska, M.; Sinha, B.; Kuczlus, T.; Karch, H. Cytolethal distending toxin from shiga toxin-producing Escherichia coli O157 causes irreversible G2/M arrest, Inhibition of proliferation, and Death of human endothelial cells. Infect. Immun. 2005, 73, 552–562.
- Taieb, F.; Nougayrède, J.; Watrin, C.; Samba-louaka, A.; Oswald, E. Escherichia coli cyclomodulin Cif induces G 2 arrest of the host cell cycle without activation of the DNA-damage checkpoint-signalling pathway. 2006, 8, 1910–1921.
- Blazkova, H.; Krejcikova, K.; Moudry, P.; Frisan, T.; Hodny, Z.; Bartek, J. Bacterial intoxication evokes cellular senescence with persistent DNA damage and cytokine signalling. J. Cell. Mol. Med. 2010, 14, 357–367.
- Liyanage, N.P.M.; Manthey, K.C.; Dassanayake, R.P.; Kuszynski, C.A.; Oakley, G.G.; Duhamel, G.E. Helicobacter hepaticus cytolethal distending toxin causes cell death in Intestinal epithelial cells via mitochondrial apoptotic pathway. Helicobacter 2010, 15, 98–107.
- Fedor, Y.; Vignard, J.; Nicolau-Travers, M.-L.; Boutet-Robinet, E.; Watrin, C.; Salles, B.; Mirey, G. From single-strand breaks to double-strand breaks during S-phase: A new mode of action of the Escherichia coli cytolethal distending toxin. Cell. Microbiol. 2013, 15, 1–15.
- Vignard, J.; Mirey, G.; Salles, B. Ionizing-radiation induced DNA double-strand breaks: A direct and indirect lighting up. Radiother. Oncol. 2013, 108, 362–369.
- Ahn, J.; Urist, M.; Prives, C. The Chk2 protein kinase. DNA Repair (Amst) 2004, 3, 1039–1047.
- Sato, T.; Koseki, T.; Yamato, K.; Saiki, K.; Konishi, K.; Yoshikawa, M.; Ishikawa, I.; Nishihara, T. p53-Independent Expression of p21 CIP1/WAF1 in plasmacytic cells during G2 cell cycle arrest induced by actinobacillus actinomycetemcomitans cytolethal distending toxin. Infect. Immun. 2002, 70, 528–534.
- Hochegger, H.; Takeda, S.; Hunt, T. Cyclin-dependent kinases and cell-cycle transitions: Does one fit all? Nat. Rev. Mol. Cell Biol. 2008, 9, 910–916.
- Langerak, P.; Russell, P. Regulatory networks integrating cell cycle control with DNA damage checkpoints and double-strand break repair. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 3562–3571.
- Alby, F.; Mazars, R.; de Rycke, J.; Guillou, E.; Baldin, V.; Darbon, J.M.; Ducommun, B. Study of the cytolethal distending toxin (CDT)-activated cell cycle checkpoint. Involvement of the CHK2 kinase. FEBS Lett. 2001, 491, 261–265.
- De Rycke, J.; Sert, V.; Comayras, C.; Tasca, C. Sequence of lethal events in HeLa cells exposed to the G2 blocking cytolethal distending toxin. Eur. J. Cell Biol. 2000, 79, 192–201.
- Escalas, N.; Davezac, N.; de Rycke, J.; Baldin, V.; Mazars, R.; Ducommun, B. Study of the cytolethal distending toxin-induced cell cycle arrest in HeLa cells: Involvement of the CDC25 phosphatase. Exp. Cell Res. 2000, 257, 206–212.
- Bielaszewska, M.; Fell, M.; Greune, L.; Prager, R.; Fruth, A.; Tschäpe, H.; Schmidt, M.A.; Karch, H.; Tscha, H. Characterization of cytolethal distending toxin genes and expression in shiga toxin-producing Escherichia coli strains of non-O157 serogroups. Infect. Immun. 2004, 72, 1812–1816.
- Cortes-Bratti, X.; Frisan, T.; Thelestam, M. The cytolethal distending toxins induce DNA damage and cell cycle arrest. Toxicon 2001, 39, 1729–1736.
- Frisan, T.; Cortes-bratti, X.; Chaves-olarte, E.; Stenerlöw, B.; Thelestam, M.; Rica, U.D.C.; José, S.; Rica, C. The Haemophilus ducreyi cytolethal distending toxin induces DNA double-strand breaks and promotes ATM-dependent activation of RhoA. Cell. Microbiol. 2003, 5, 695–707.
- Abbas, T.; Dutta, A. P21 in Cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414.
- Shenker, B.J.; Hoffmaster, R.H.; Zekavat, A.; Yamaguchi, N.; Lally, E.T.; Demuth, D.R. Induction of apoptosis in human T cells by actinobacillus actinomycetemcomitans cytolethal distending toxin Is a consequence of G2 arrest of the cell cycle. J. Immunol. 2001, 167, 435–441.
- Ohara, M.; Hayashi, T.; Kusunoki, Y.; Miyauchi, M.; Takata, T.; Sugai, M.; Mmun, I.N.I. Caspase-2 and Caspase-7 are involved in cytolethal distending toxin-induced apoptosis in jurkat and MOLT-4 T-cell lines. Infect. Immun. 2004, 72, 871–879.
- Hickey, T.E.; Majam, G.; Guerry, P. Intracellular survival of campylobacter jejuni in human monocytic cells and induction of apoptotic death by cytholethal distending toxin. Infect. Immun. 2005, 73, 5194–5197.
- Guidi, R.; Guerra, L.; Levi, L.; Stenerlöw, B.; Fox, J.G.; Josenhans, C.; Masucci, M.G.; Frisan, T. Chronic exposure to the cytolethal distending toxins of Gram-negative bacteria promotes genomic instability and altered DNA damage response. Cell. Microbiol. 2013, 15, 98–113.
- Gelfanova, V.; Hansen, E.J.; Spinola, S.M. Cytolethal distending toxin of haemophilus ducreyi induces apoptotic death of jurkat T cells. Infect. Immun. 1999, 67, 6394–6402.
- Wising, C.; Azem, J.; Zetterberg, M.; Svensson, L.A.; Ahlman, K.; Lagerga, T. Induction of apoptosis/necrosis in various human cell lineages by haemophilus ducreyi cytolethal distending toxin. Toxicon 2005, 45, 767–776.
- Shenker, B.J.; Dlakic, M.; Walker, L.P.; Besack, D.; Jaffe, E.; LaBelle, E.; Boesze-Battaglia, K. A novel mode of action for a microbial-derivated immunotoxin: The cytolethal distending toxin subunit B exhibits phosphatidylinositol3,4,5-triphosphate phosphatase activity. J. Immunol. 2007, 178, 5099–5108.
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705.
- Guerra, L.; Guidi, R.; Frisan, T. Do bacterial genotoxins contribute to chronic inflammation, genomic instability and tumor progression? FEBS J. 2011, 278, 4577–4588.