 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Luciana Marti | + 2956 word(s) | 2956 | 2021-05-28 06:02:11 | | | |
| 2 | Enzi Gong | Meta information modification | 2956 | 2021-06-01 10:07:11 | | | | |
| 3 | Enzi Gong | Meta information modification | 2956 | 2021-06-03 05:45:00 | | | | |
| 4 | Enzi Gong | + 3022 word(s) | 5978 | 2021-06-03 05:53:34 | | | | |
| 5 | Enzi Gong | + 3022 word(s) | 5978 | 2021-06-03 05:59:44 | | | | |
| 6 | Enzi Gong | -3022 word(s) | 2956 | 2021-06-03 06:03:44 | | | | |
| 7 | Enzi Gong | -3088 word(s) | 2890 | 2021-06-03 06:09:09 | | | | |
| 8 | Enzi Gong | -3088 word(s) | 2890 | 2021-06-03 06:10:21 | | | | |
| 9 | Enzi Gong | Meta information modification | 2890 | 2021-06-03 07:31:44 | | | | |
| 10 | Enzi Gong | Meta information modification | 2890 | 2021-06-03 07:45:44 | | | | |
| 11 | Enzi Gong | + 61 word(s) | 2951 | 2021-06-21 08:50:55 | | |
Video Upload Options
Lymph node structural organization is reported to be governed by the stromal cells. Fibroblast reticular cells (FRCs), a subset of the stromal cells found in the T lymphocyte region of lymph nodes (LNs) and other secondary lymphoid organs (SLOs), have been described as much more than structural cells.
FRCs are described to be organized in a conduit system called the “reticular fiber network”, responsible for transferring antigens from tissue to T cell zones in LNs and for controlling the conduit matrix deposition during lymph node expansion.
1. Overview
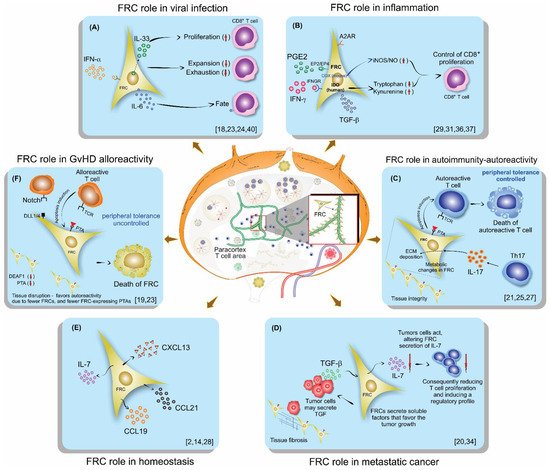
Fibroblastic reticular cells (FRCs), usually found and isolated from the T cell zone of lymph nodes, have recently been described as much more than simple structural cells. Originally, these cells were described to form a conduit system called the “reticular fiber network” and for being responsible for transferring the lymph fluid drained from tissues through afferent lymphatic vessels to the T cell zone. However, nowadays, these cells are described as being capable of secreting several cytokines and chemokines and possessing the ability to interfere with the immune response, improving it, and also controlling lymphocyte proliferation. Here, we performed a systematic review of the several methods employed to investigate the mechanisms used by fibroblastic reticular cells to control the immune response, as well as their ability in determining the fate of T cells. We searched articles indexed and published in the last five years, between 2016 and 2020, in PubMed, Scopus, and Cochrane, following the PRISMA guidelines. We found 175 articles published in the literature using our searching strategies, but only 24 articles fulfilled our inclusion criteria and are discussed here. Other articles important in the built knowledge of FRCs were included in the introduction and discussion. The studies selected for this review used different strategies in order to access the contribution of FRCs to different mechanisms involved in the immune response: 21% evaluated viral infection in this context, 13% used a model of autoimmunity, 8% used a model of GvHD or cancer, 4% used a model of Ischemic-reperfusion injury (IRI). Another four studies just targeted a particular signaling pathway, such as MHC II expression, FRC microvesicles, FRC secretion of IL-15, FRC network, or ablation of the lysophosphatidic acid (LPA)-producing ectoenzyme autotaxin. In conclusion, our review shows the strategies used by several studies to isolate and culture fibroblastic reticular cells, the models chosen by each one, and dissects their main findings and implications in homeostasis and disease.
2. Background
3. Conclusions
References
- Denton, A.E.; Carr, E.J.; Magiera, L.P.; Watts, A.J.B.; Fearon, D.T. Embryonic FAP+ lymphoid tissue organizer cells generate the reticular network of adult lymph nodes. J. Exp. Med. 2019, 216, 2242–2252.
- Link, A.; Vogt, T.K.; Favre, S.; Britschgi, M.R.; Acha-Orbea, H.; Hinz, B.; Cyster, J.G.; Luther, S.A. Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive T cells. Nat. Immunol. 2007, 8, 1255–1265.
- Sixt, M.; Kanazawa, N.; Selg, M.; Samson, T.; Roos, G.; Reinhardt, D.P.; Pabst, R.; Lutz, M.B.; Sorokin, L. The conduit system transports soluble antigens from the afferent lymph to resident dendritic cells in the T cell area of the lymph node. Immunity 2005, 22, 19–29.
- Martinez, V.G.; Pankova, V.; Krasny, L.; Singh, T.; Makris, S.; White, I.J.; Benjamin, A.C.; Dertschnig, S.; Horsnell, H.L.; Kriston-Vizi, J.; et al. Fibroblastic Reticular Cells Control Conduit Matrix Deposition during Lymph Node Expansion. Cell Rep. 2019, 29, 2810–2822.
- Luther, S.A.; Vogt, T.K.; Siegert, S. Guiding blind T cells and dendritic cells: A closer look at fibroblastic reticular cells found within lymph node T zones. Immunol. Lett. 2011, 138, 9–11.
- Mueller, S.N.; Ahmed, R. Lymphoid stroma in the initiation and control of immune responses. Immunol. Rev. 2008, 224, 284–294.
- Alvarenga, H.G.; Marti, L. Multifunctional Roles of Reticular Fibroblastic Cells: More Than Meets the Eye? J. Immunol. Res. 2014, 2014, 402038.
- Fletcher, A.L.; Acton, S.E.; Knoblich, K. Lymph node fibroblastic reticular cells in health and disease. Nat. Rev. Immunol. 2015, 15, 350–361.
- Severino, P.; Palomino, D.T.; Alvarenga, H.; Almeida, C.B.; Pasqualim, D.C.; Cury, A.; Salvalaggio, P.R.; De Vasconcelos Macedo, A.L.; Andrade, M.C.; Aloia, T.; et al. Human Lymph Node-Derived Fibroblastic and Double-Negative Reticular Cells Alter Their Chemokines and Cytokines Expression Profile Following Inflammatory Stimuli. Front. Immunol. 2017, 8, 141.
- Vega, F.; Coombes, K.R.; Thomazy, V.A.; Patel, K.; Lang, W.; Jones, D. Tissue-specific function of lymph node fibroblastic reticulum cells. Pathobiol. J. Immunopathol. Mol. Cell. Biol. 2006, 73, 71–81.
- Frontera, V.; Arcangeli, M.L.; Zimmerli, C.; Bardin, F.; Obrados, E.; Audebert, S.; Bajenoff, M.; Borg, J.P.; Aurrand-Lions, M. Cutting edge: JAM-C controls homeostatic chemokine secretion in lymph node fibroblastic reticular cells expressing thrombomodulin. J. Immunol. 2011, 187, 603–607.
- Kaldjian, E.P.; Gretz, J.E.; Anderson, A.O.; Shi, Y.; Shaw, S. Spatial and molecular organization of lymph node T cell cortex: A labyrinthine cavity bounded by an epithelium-like monolayer of fibroblastic reticular cells anchored to basement membrane-like extracellular matrix. Int. Immunol. 2001, 13, 1243–1253.
- Lukacs-Kornek, V.; Malhotra, D.; Fletcher, A.L.; Acton, S.E.; Elpek, K.G.; Tayalia, P.; Collier, A.-r.; Turley, S.J. Regulated release of nitric oxide by nonhematopoietic stroma controls expansion of the activated T cell pool in lymph nodes. Nat. Immunol. 2011, 12, 1096–1104.
- Fletcher, A.L.; Malhotra, D.; Turley, S.J. Lymph node stroma broaden the peripheral tolerance paradigm. Trends Immunol. 2011, 32, 12–18.
- Gardner, J.M.; Devoss, J.J.; Friedman, R.S.; Wong, D.J.; Tan, Y.X.; Zhou, X.; Johannes, K.P.; Su, M.A.; Chang, H.Y.; Krummel, M.F.; et al. Deletional tolerance mediated by extrathymic Aire-expressing cells. Science 2008, 321, 843–847.
- Nadafi, R.; Gago de Graça, C.; Keuning, E.D.; Koning, J.J.; de Kivit, S.; Konijn, T.; Henri, S.; Borst, J.; Reijmers, R.M.; van Baarsen, L.G.M.; et al. Lymph Node Stromal Cells Generate Antigen-Specific Regulatory T Cells and Control Autoreactive T and B Cell Responses. Cell Rep. 2020, 30, 4110–4123.
- Krausgruber, T.; Fortelny, N.; Fife-Gernedl, V.; Senekowitsch, M.; Schuster, L.C.; Lercher, A.; Nemc, A.; Schmidl, C.; Rendeiro, A.F.; Bergthaler, A.; et al. Structural cells are key regulators of organ-specific immune responses. Nature 2020, 583, 296–302.
- Aparicio-Domingo, P.; Cannelle, H.; Buechler, M.B.; Nguyen, S.; Kallert, S.M.; Favre, S.; Alouche, N.; Papazian, N.; Ludewig, B.; Cupedo, T.; et al. Fibroblast-derived IL-33 is dispensable for lymph node homeostasis but critical for CD8 T-cell responses to acute and chronic viral infection. Eur. J. Immunol. 2020, 51, 76–90.
- Dertschnig, S.; Evans, P.; Santos, E.S.P.; Manzo, T.; Ferrer, I.R.; Stauss, H.J.; Bennett, C.L.; Chakraverty, R. Graft-versus-host disease reduces lymph node display of tissue-restricted self-antigens and promotes autoimmunity. J. Clin. Investig. 2020, 130, 1896–1911.
- Eom, J.; Park, S.M.; Feisst, V.; Chen, C.J.J.; Mathy, J.E.; McIntosh, J.D.; Angel, C.E.; Bartlett, A.; Martin, R.; Mathy, J.A.; et al. Distinctive Subpopulations of Stromal Cells Are Present in Human Lymph Nodes Infiltrated with Melanoma. Cancer Immunol. Res. 2020, 8, 990–1003.
- Gonzalez Badillo, F.; Zisi Tegou, F.; Masina, R.; Wright, S.; Scully, M.; Harwell, L.; Lupp, M.; Postigo-Fernandez, J.; Creusot, R.J.; Tomei, A.A. Tissue-Engineered Stromal Reticula to Study Lymph Node Fibroblastic Reticular Cells in Type I Diabetes. Cell. Mol. Bioeng. 2020, 13, 419–434.
- Knop, L.; Deiser, K.; Bank, U.; Witte, A.; Mohr, J.; Philipsen, L.; Fehling, H.J.; Müller, A.J.; Kalinke, U.; Schüler, T. IL-7 derived from lymph node fibroblastic reticular cells is dispensable for naive T cell homeostasis but crucial for central memory T cell survival. Eur. J. Immunol. 2020, 50, 846–857.
- Perez-Shibayama, C.; Islander, U.; Lütge, M.; Cheng, H.W.; Onder, L.; Ring, S.S.; de Martin, A.; Novkovic, M.; Colston, J.; Gil-Cruz, C.; et al. Type I interferon signaling in fibroblastic reticular cells prevents exhaustive activation of antiviral CD8+ T cells. Sci. Immunol. 2020, 5.
- Brown, F.D.; Sen, D.R.; LaFleur, M.W.; Godec, J.; Lukacs-Kornek, V.; Schildberg, F.A.; Kim, H.J.; Yates, K.B.; Ricoult, S.J.H.; Bi, K.; et al. Fibroblastic reticular cells enhance T cell metabolism and survival via epigenetic remodeling. Nat. Immunol. 2019, 20, 1668–1680.
- Kasinath, V.; Yilmam, O.A.; Uehara, M.; Jiang, L.; Ordikhani, F.; Li, X.; Salant, D.J.; Abdi, R. Activation of fibroblastic reticular cells in kidney lymph node during crescentic glomerulonephritis. Kidney Int. 2019, 95, 310–320.
- Kelch, I.D.; Bogle, G.; Sands, G.B.; Phillips, A.R.J.; LeGrice, I.J.; Dunbar, P.R. High-resolution 3D imaging and topological mapping of the lymph node conduit system. PLoS Biol. 2019, 17, e3000486.
- Majumder, S.; Amatya, N.; Revu, S.; Jawale, C.V.; Wu, D.; Rittenhouse, N.; Menk, A.; Kupul, S.; Du, F.; Raphael, I.; et al. IL-17 metabolically reprograms activated fibroblastic reticular cells for proliferation and survival. Nat. Immunol. 2019, 20, 534–545.
- Masters, A.R.; Hall, A.; Bartley, J.M.; Keilich, S.R.; Lorenzo, E.C.; Jellison, E.R.; Puddington, L.; Haynes, L. Assessment of Lymph Node Stromal Cells as an Underlying Factor in Age-Related Immune Impairment. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2019, 74, 1734–1743.
- Schaeuble, K.; Cannelle, H.; Favre, S.; Huang, H.Y.; Oberle, S.G.; Speiser, D.E.; Zehn, D.; Luther, S.A. Attenuation of chronic antiviral T-cell responses through constitutive COX2-dependent prostanoid synthesis by lymph node fibroblasts. PLoS Biol. 2019, 17, e3000072.
- Dubrot, J.; Duraes, F.V.; Harlé, G.; Schlaeppi, A.; Brighouse, D.; Madelon, N.; Göpfert, C.; Stokar-Regenscheit, N.; Acha-Orbea, H.; Reith, W.; et al. Absence of MHC-II expression by lymph node stromal cells results in autoimmunity. Life Sci. Alliance 2018, 1, e201800164.
- Knoblich, K.; Cruz Migoni, S.; Siew, S.M.; Jinks, E.; Kaul, B.; Jeffery, H.C.; Baker, A.T.; Suliman, M.; Vrzalikova, K.; Mehenna, H.; et al. The human lymph node microenvironment unilaterally regulates T-cell activation and differentiation. PLoS Biol. 2018, 16, e2005046.
- Maarouf, O.H.; Uehara, M.; Kasinath, V.; Solhjou, Z.; Banouni, N.; Bahmani, B.; Jiang, L.; Yilmam, O.A.; Guleria, I.; Lovitch, S.B.; et al. Repetitive ischemic injuries to the kidneys result in lymph node fibrosis and impaired healing. JCI Insight 2018, 3.
- Chung, J.; Ebens, C.L.; Perkey, E.; Radojcic, V.; Koch, U.; Scarpellino, L.; Tong, A.; Allen, F.; Wood, S.; Feng, J.; et al. Fibroblastic niches prime T cell alloimmunity through Delta-like Notch ligands. J. Clin. Investig. 2017, 127, 1574–1588.
- Gao, J.; Zhao, L.; Liu, L.; Yang, Y.; Guo, B.; Zhu, B. Disrupted fibroblastic reticular cells and interleukin-7 expression in tumor draining lymph nodes. Oncol. Lett. 2017, 14, 2954–2960.
- Pasztoi, M.; Pezoldt, J.; Beckstette, M.; Lipps, C.; Wirth, D.; Rohde, M.; Paloczi, K.; Buzas, E.I.; Huehn, J. Mesenteric lymph node stromal cell-derived extracellular vesicles contribute to peripheral de novo induction of Foxp3(+) regulatory T cells. Eur. J. Immunol. 2017, 47, 2142–2152.
- Valencia, J.; Jiménez, E.; Martínez, V.G.; Del Amo, B.G.; Hidalgo, L.; Entrena, A.; Fernández-Sevilla, L.M.; Del Río, F.; Varas, A.; Vicente, Á.; et al. Characterization of human fibroblastic reticular cells as potential immunotherapeutic tools. Cytotherapy 2017, 19, 640–653.
- Yu, M.; Guo, G.; Zhang, X.; Li, L.; Yang, W.; Bollag, R.; Cui, Y. Fibroblastic reticular cells of the lymphoid tissues modulate T cell activation threshold during homeostasis via hyperactive cyclooxygenase-2/prostaglandin E(2) axis. Sci. Rep. 2017, 7, 3350.
- Gil-Cruz, C.; Perez-Shibayama, C.; Onder, L.; Chai, Q.; Cupovic, J.; Cheng, H.W.; Novkovic, M.; Lang, P.A.; Geuking, M.B.; McCoy, K.D.; et al. Fibroblastic reticular cells regulate intestinal inflammation via IL-15-mediated control of group 1 ILCs. Nat. Immunol. 2016, 17, 1388–1396.
- Novkovic, M.; Onder, L.; Cupovic, J.; Abe, J.; Bomze, D.; Cremasco, V.; Scandella, E.; Stein, J.V.; Bocharov, G.; Turley, S.J.; et al. Topological Small-World Organization of the Fibroblastic Reticular Cell Network Determines Lymph Node Functionality. PLoS Biol. 2016, 14, e1002515.
- Royer, D.J.; Conrady, C.D.; Carr, D.J. Herpesvirus-Associated Lymphadenitis Distorts Fibroblastic Reticular Cell Microarchitecture and Attenuates CD8 T Cell Responses to Neurotropic Infection in Mice Lacking the STING-IFNα/β Defense Pathways. J. Immunol. 2016, 197, 2338–2352.
- Takeda, A.; Kobayashi, D.; Aoi, K.; Sasaki, N.; Sugiura, Y.; Igarashi, H.; Tohya, K.; Inoue, A.; Hata, E.; Akahoshi, N.; et al. Fibroblastic reticular cell-derived lysophosphatidic acid regulates confined intranodal T-cell motility. eLife 2016, 5, e10561.
- Krishnamurty, A.T.; Turley, S.J. Lymph node stromal cells: Cartographers of the immune system. Nat. Immunol. 2020, 21, 369–380.
- Talemi, S.R.; Höfer, T. Antiviral interferon response at single-cell resolution. Immunol. Rev. 2018, 285, 72–80.
- Hunter, C.A.; Jones, S.A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015, 16, 448–457.
- Chai, Q.; Onder, L.; Scandella, E.; Gil-Cruz, C.; Perez-Shibayama, C.; Cupovic, J.; Danuser, R.; Sparwasser, T.; Luther, S.A.; Thiel, V.; et al. Maturation of lymph node fibroblastic reticular cells from myofibroblastic precursors is critical for antiviral immunity. Immunity 2013, 38, 1013–1024.
- Thompson, H.L.; Smithey, M.J.; Uhrlaub, J.L.; Jeftić, I.; Jergović, M.; White, S.E.; Currier, N.; Lang, A.M.; Okoye, A.; Park, B.; et al. Lymph nodes as barriers to T-cell rejuvenation in aging mice and nonhuman primates. Aging Cell 2019, 18, e12865.
- Mueller, S.N.; Matloubian, M.; Clemens, D.M.; Sharpe, A.H.; Freeman, G.J.; Gangappa, S.; Larsen, C.P.; Ahmed, R. Viral targeting of fibroblastic reticular cells contributes to immunosuppression and persistence during chronic infection. Proc. Natl. Acad. Sci. USA 2007, 104, 15430–15435.
- Zeng, M.; Smith, A.J.; Wietgrefe, S.W.; Southern, P.J.; Schacker, T.W.; Reilly, C.S.; Estes, J.D.; Burton, G.F.; Silvestri, G.; Lifson, J.D.; et al. Cumulative mechanisms of lymphoid tissue fibrosis and T cell depletion in HIV-1 and SIV infections. J. Clin. Investig. 2011, 121, 998–1008.
- Malhotra, D.; Fletcher, A.L.; Turley, S.J. Stromal and hematopoietic cells in secondary lymphoid organs: Partners in immunity. Immunol. Rev. 2013, 251, 160–176.
- Siegert, S.; Luther, S.A. Positive and negative regulation of T cell responses by fibroblastic reticular cells within paracortical regions of lymph nodes. Front. Immunol. 2012, 3, 285.
- Postigo-Fernandez, J.; Farber, D.L.; Creusot, R.J. Phenotypic alterations in pancreatic lymph node stromal cells from human donors with type 1 diabetes and NOD mice. Diabetologia 2019, 62, 2040–2051.


