 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Viorica Patrulea | + 13497 word(s) | 13497 | 2020-09-07 11:21:13 | | | |
| 2 | Dean Liu | -563 word(s) | 12934 | 2020-12-09 02:23:09 | | |
Video Upload Options
Antimicrobial peptides (AMPs), also called host defense peptides (HDPs), are found in bacteria, fungi, plants, and animals. They typically consist of 10–50 amino acid residues (very rarely up to 100 amino acids) and generally possess cationic (net charge ranging from −4 to +20) and amphipathic structures.
1. Introduction
Skin wound healing is a complex and highly orchestrated process that consists of four overlapping stages, namely inflammation, proliferation, migration, and the maturation of the new tissue [1][2]. If any of these phases is disturbed, healing may lead to either chronic non-healing wounds, such as venous, diabetic, and pressure ulcers or pathological scaring and excessive fibrosis. Major factors leading to chronic wounds are ischemia and microbial colonization [3][4]. These types of wound infections may serve as a bacterial reservoir and cause high morbidity and costs [5]. Nowadays, microbial infections due to multidrug resistance (MDR) are an important threat, which needs to be seriously considered. MDR bacteria hinder wound healing, as most of the wounds would develop infections at some point. The most opportunistic and MDR pathogens, which have the ability to colonize the wound, are included in the ESKAPE bacterial collection (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumonia, Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacter species) [6]. S. aureus and P. aeruginosa have additionally the ability to develop a biofilm on the wound and medical devices, which worsens the situation and makes biofilm bacteria extremely difficult or even impossible to treat. This is attributed to the fact that bacteria within biofilms are 100 to 1000-fold more tolerant to antimicrobial agents, thus delaying the healing of infected wounds [7]. Additionally, bacteria within biofilms are enclosed in a 3D network of a self-produced matrix of extracellular polymeric substances (EPS), such as exopolysaccharides, proteins, extracellular DNA, and teichoic and lipoteichoic acids, which amplifies the resistance to most of the existing antimicrobial agents [8]. Infected wounds are an alarming cause of death, especially in immunocompromised and diabetic patients, which poses a significant clinical and economic burden for the patients and the healthcare system.
In these contexts, antimicrobial peptides (AMPs) are used as promising alternatives to counter bacterial infections and control microbial spreading. As antimicrobial resistance (AMR) is currently becoming a worldwide threat, the main focuses of research are: (i) to identify existing or new AMPs and to characterize their efficacy against prevalent microorganisms, including their mechanism of action and toxicity toward mammalian cells and/or blood cells; (ii) to assess the role of AMPs in modulating the pro-inflammatory cytokines and in adaptive immune mechanisms; and (iii) to identify new approaches for AMPs delivery [9].
The main goal of this review is to identify and discuss potent AMPs that are able to eradicate MDR bacteria in wound infections and AMPs acting as wound-healing promotors. AMP dendrimers (AMPDs), similar to AMPs, are very potent against the ESKAPE collection and biofilm-related bacteria; these are discussed as well. We also review strategies to deliver AMPs topically—their covalent coupling, self-assembly, coupling to antibiotics, embedding into 3D scaffold or dressing, the use of nanotechnological platforms (i.e., organic or inorganic nanoparticles) and smart nanomaterials—as future promising technologies to deliver AMPs efficiently to the site of infection, and CRISPR-Cas are also discussed.
2. Antimicrobial Peptides (AMPs) and AMP Dendrimers (AMPDs)
AMPs are small molecules having a broad activity to treat microbial infections, especially those causing AMR [10]. They have several advantages over current antibiotic treatment, which we will further detailed and which makes them good candidates especially for the treatment of topical and systemic infections. The selection of AMPs for the treatment of different infectious diseases may start with an in vitro/in silico screening to determine their biological activity against different microbial strains. An in silico approach refers to the bioinformatics tools that use different algorithms to classify, predict, and design new AMPs [11][12]. The rational of in silico design relies on its potential to drastically reduce the costs for AMPs production and the time to evaluate their biological activity and potential toxicity by predicting in advance the bioactivity of the AMPs [6]. This methodology was shown to have a 94% accuracy in terms of biological activity and could predict which AMPs would be active in vitro [13]. Kumar Meher et al. developed a support vector machine (SVM)-based computational approach for the prediction of AMPs with an improved accuracy [14]. The most advanced computational tools to predict the biological activity of the AMPs with the best accuracy have been described in depth by different authors in the literature [12][15][16][17]. These “tailored” AMPs could help overcome microbial resistance and improve pathogen killing.
2.1. AMPs: Classification, Mechanism of Action
Antimicrobial peptides (AMPs), also called host defense peptides (HDPs), are found in bacteria, fungi, plants, and animals. They typically consist of 10–50 amino acid residues (very rarely up to 100 amino acids) and generally possess cationic (net charge ranging from −4 to +20) and amphipathic structures. They were identified for the first time in 1922, almost a century ago, by Alexander Fleming, who discovered lysozyme [18], an antibacterial enzyme found in saliva, tears, and human urine. Soon after, in 1928, he discovered penicillin, the first antibiotic in human history, which was extracted from the culture of green mold, Penicillium notatum [19]. Since then, more than 3171 AMPs have been reported according to the Antimicrobial Peptide Database (APD) [20]. According to the DRAMP (data repository of antimicrobial peptides, an open access and manually annotated database), there are currently 20,227 general entries related to AMPs, of which 5412 are general AMPs (including natural and synthetic AMPs), 14,739 entries are patented AMPs, and 76 are AMPs that are under development as drugs (preclinical or clinical phase), most of them having antimicrobial activity [21]. The main focus of the DRAMP database is to survey the hemolytic activity of AMPs, including detailed test protocols essential to evaluate the efficacy/toxicity balance of the AMPs [22].
Based on their origin, data source, activity against microorganisms, taxonomy, conformational structure, and amino acid composition, AMPs can be divided into several categories, as depicted in Figure 1. Very often, AMPs are classified based on their secondary conformation into four different families: α-helix, β-sheet, loop, and extended family. Most of the AMPs fall into the α-helix (e.g., cryptdin-4, human α-defensins (HD-5 and 6), magainin 1 and 2, melittin, moricin) or β-sheet families (e.g., human β-defensins (hBD-1-6), lactoferricin B, protegrin-1, tachyplesin I) [23]. Very few AMPs show a loop structure (e.g., thanatin), and extended structures with neither α-helical nor β-sheet conformation (e.g., Indolicidin, Indolicidin analogue (CP10A), tritrpticin) [6].
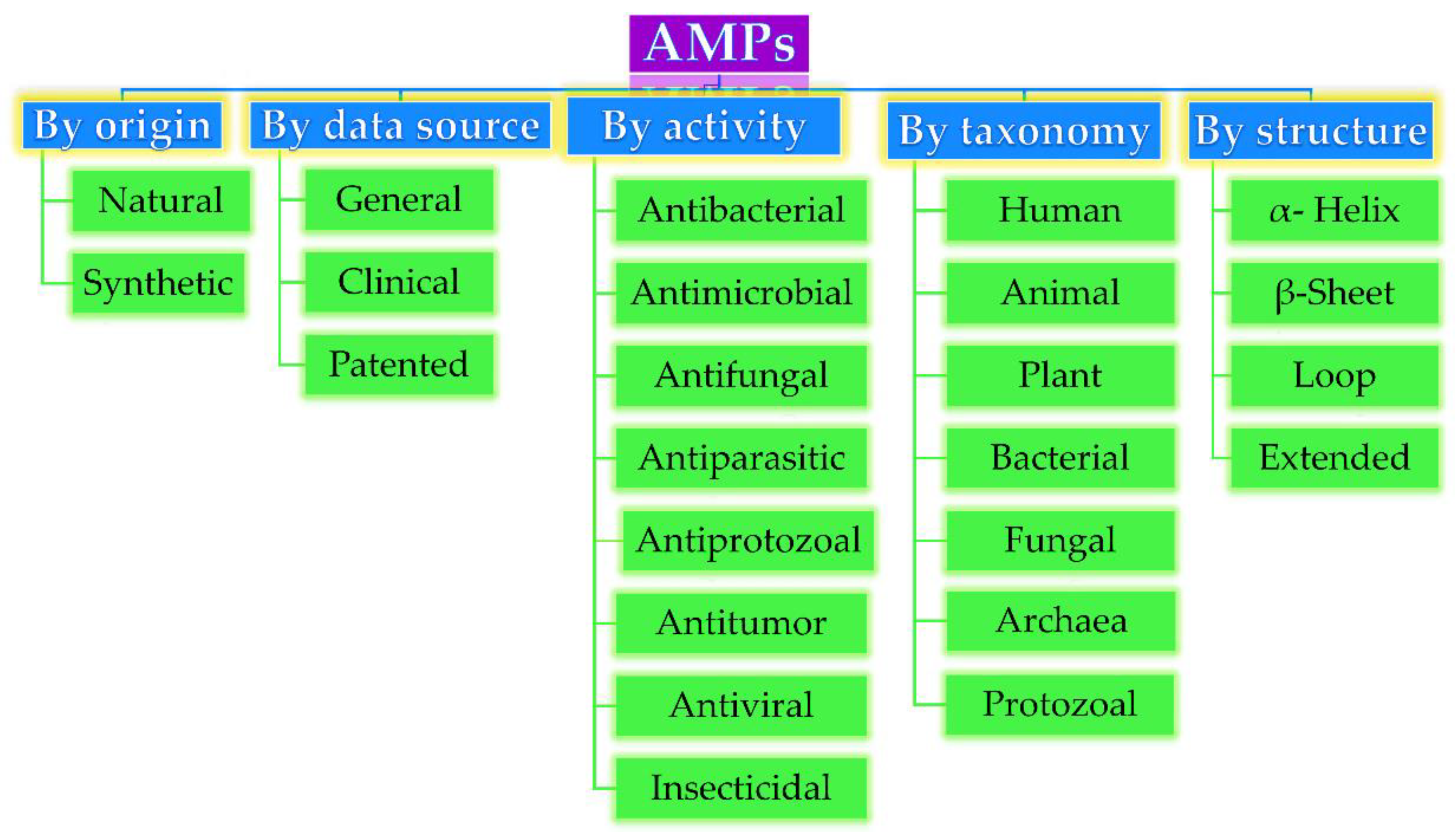 Figure 1. Antimicrobial peptides (AMP) classification (partially inspired by the DRAMP database [21]). DRAMP: data repository of antimicrobial peptides, an open access and manually annotated database.
Figure 1. Antimicrobial peptides (AMP) classification (partially inspired by the DRAMP database [21]). DRAMP: data repository of antimicrobial peptides, an open access and manually annotated database.
The biological activity of AMPs against both Gram-positive (Gram (+)) and Gram-negative (Gram (−)) bacteria, viruses, and fungi has drawn special attention, particularly their ability to kill MDR bacteria [24]. AMPs act mostly by disrupting the integrity of the cell membrane, which is accompanied by the leakage of vesicles and other cellular components within a very short time. Typically, their interaction is based on the electrostatic attraction between the negatively charged bacterial wall and positively charged AMPs [25]. Subsequently, once the interaction is established, AMPs access the phospholipid bilayer membrane and start to aggregate, leading to the formation of different complex structures through at least four distinct mechanisms, such as the formation of “aggregate”, “toroidal pore”, “barrel-state”, or “carpet” models, or via membrane permeabilization that would lead finally to cell lysis. These mechanisms are described in detail by Rios et al. [10] and Thapa et al. [3]. In addition to these bilayer membrane mechanisms, AMPs can diffuse via cytoplasmic membrane and accumulate intracellularly, subsequently blocking DNA replication and disrupting RNA and protein synthesis, which in turn leads to cell wall lysis. Buforin II, pleurocidin, and dermaseptin are examples of those [26]. A recent study showed that some AMPs such as cathelicidins [27] and papiliocin [28] can act via the production of reactive oxygen species (ROS) and mitochondrial dysfunction, thus leading to cell apoptosis.
2.1.1. Wound-Healing Promoting AMPs
AMPs not only exhibit a broad spectrum of antibacterial activity, but many of them display antibiofilm, anti-methicillin-resistant Staphylococcus aureus (MRSA), anti-tuberculosis, anti-sepsis, anti-toxin, antiviral, anti-HIV, antifungal, antiparasitic, anticancer, anti-diabetic, wound healing and anti-inflammatory activities as well. Some examples of AMPs split according to their activity are listed in Table 1.
Table 1. AMPs with their potential activity selected from the Antimicrobial Peptide Database (APD) [20].
|
Activity AMP |
Selected Examples of AMPs |
Total No. |
|
Antibacterial peptides |
Abaecin; andropin; bombinin; 1 bBD-1-13; cecropin A, B, C, D, P; cryptdin; drosocin; esculentin-1-2; dermaseptin-B2-B5, B6, S1-S4; 2 hBD-26,27; LL-37; magainin; melittin; nisin; protegrin 1; pyrrhocoricin; temporin A, B, C, E, F, G, K, L; thanatin; tritrpticin |
2678 |
|
Antibiofilm peptides |
BMAP-27,28; citropin 1.1; colistin A; Dhvar4; gramicidin S; hBD-3; holothuroidin 1; indolicidin; LL-37; nisin A; polymyxin B; protegrin 1; SMAP-29 (Ovispirin); tachyplesin III; temporin B; temporin-1CEb
|
57 |
|
Anticancer peptides |
Alloferon 1,2; aurein 1–3; buforin II; gomesin; indolicidin; lactoferricin B; LL-37; magainin 2; mastoparan B; melittin; nisin A,Z; tritrpticin |
237 |
|
Anti-diabetic peptides |
Amolopin; brevinin-1E, 2EC; esculentin-1, 1B; magainin-AM2 |
15 |
|
Antifungal peptides |
Androctonin; antifungal protein; aurein 1–3; cecropin 2, A, B; dermaseptin-S1-S5; HD-2-6; HNP-1-6; indolicidin; lactoferricin B; magainin 2; melittin; protegrin 1–5; ponericin G1-G4; G7, W1-W5; thanatin; tritrpticin |
1142 |
|
Anti-HIV peptides |
Aurein 1.2; cecropin A; dermaseptin-S1,S4, S9; hBD-2,3; HNP-1-4; indolicidin; lactoferricin B; LL-37; melittin; protegrin 1 |
109 |
|
Anti-inflammatory peptides |
Allomyrinasin; cathelicidin-PY; coprisin; defensin DEFB126; lucilin; papiliocin |
20 |
|
Anti-MRSA peptides |
Acipensin 1,2; BMAP-27,28; CAP18, citropin 1.1; clavanin A; cryptdin-4; 4 Dhvar5; esculentin-1,2 ISa-ISb; hBD-3; hedistin; 3 HNP-1; hominicin; imcroporin; indolicidin; LL-37; micasin-1; omega76; SMAP-29; plectasin; pleurocidin; protegrin 1; ubiquicidin |
165 |
|
Antiparasitic peptides |
Batroxicidin; cecropin A; dermaseptin-S1-S5; kalata B2, B5-B7; LL-37; magainin 2; melittin; temporin A, B, F, L; |
116 |
|
Anti-sepsis peptides |
Apidaecin IA; bactenecin 7; buforin II; cathelicidin-PY; cecropin 2, P1; drosocin; 5 HD-5; HNP-1; lactoferricin B; LL-37; melittin; polymyxin B; protegrin 1; pyrrhocoricin; SMAP-29; tachyplesin I; temporin L; thanatin |
75 |
|
Anti-toxin peptides |
hBD-1-4; HNP-1-5; retrocyclin-1-3 |
15 |
|
Anti-tuberculosis peptides |
Griselimycin; hBD consensus; hBD10; human granulysin; lassomycin; laterosporulin10; LL-37; micrococcin P1; pantocin wh-1; RNase 7; Teixobactin; VpAmp1.0, 2.0 |
13 |
|
Antiviral peptides |
Alloferon 1,2; antiviral protein Y3; aurein 1.2; BMAP-27,28; dermaseptin-S1, S4; hBD-1-3; HNP-1-6; indolicidin; lactoferricin B; LL-37; magainin 2; melittin; mucroporin; protegrin 1–5; thanatin; temporin A,B |
189 |
|
Wound-healing peptides |
AH90; AG-30; AG-30/5C; bactenecin; coprisin; epinecidin-1; hBD-2, 3; HD-5; HNP-1; IDR-1018; indolicidin; LL-37; lucifensin; magainin 2; nisin A; temporin A |
22 |
1bBD: bovine beta defensin; 2hBD: human beta-defensin; 3HNP: human neutrophil peptide; 4Dhvar: human Histatin; 5HD: human defensin.
Many AMPs, beside their antimicrobial activity, additionally promote wound healing and stimulate angiogenesis, which are key factors in the process of tissue regeneration. Several AMPs that have shown promising results after topical application, both in vitro and in vivo, are mentioned in Table 1. For instance, LL-37 (phase II in clinical trials) was shown to promote angiogenesis, migration, and the proliferation of dermal cells, which is a crucial factor in wound restoration [29] and used for treatment of diabetic foot ulcer (DFU). IDR-1018, an innate defense regulator peptide and less toxic than LL-37, was shown to significantly accelerate wound healing in non-infected and non-diabetic murine and S. aureus-infected porcine models [30]. Human β-defensin (hBD-2) induces keratinocyte migration, while hBD-3 has been additionally reported to accelerate wound closure with a 10-fold bacterial reduction in a porcine model of S. aureus-infected diabetic wounds [31]. Furthermore, hBD-3 and LL-37 were shown to stimulate not only dermal cells, but also corneal epithelial cells with an enhanced ocular surface healing [32]. LL-37 induced the migration, proliferation, and wound closure of airway epithelial cells [33]. Pexiganan, an analogue of magainin 2 and the first AMP evaluated for human skin infections, was shown to promote dermal cell migration and antibacterial activity against Gram (+) and Gram (−) cells (minimal inhibitory concentration (MIC) ≤16 µg/mL) in diabetic foot ulcer models early in clinical trials. Consequently, Locilex®, a 0.8% pexiganan cream, was patented and developed for DFU treatment. Locilex® failed later during phase III of clinical trials due to a lack of efficacy compared to placebo or “vehicle” cream without pexiganan [34].
The lack of stability of AMPs is an important issue, and most of the AMPs could self-assemble, losing their activity even in a saline solution. Furthermore, they may be sensitive to enzymatic degradation [35]. For instance, Nishikawa et al. developed a helical AMP, AG-30, which has angiogenic properties in a mouse ischemic limb model. AG-30 exhibited high antimicrobial activity against P. aeruginosa (MIC: 5 µg/mL), E. coli (MIC: 40 µg/mL), and S. aureus (MIC: 20 µg/mL) via a membrane disruption mechanism [36]; however, it was lacking stability in saline. An improved version of AG-30, in terms of stability, is the cationic AG-30/5C, which was developed by replacing five residues of AG-30 with five cationic amino acids. It is noteworthy that AG-30/5C was shown to be stable in a saline solution for at least 12 months at 5 °C. In vitro, AG-30/5C showed a strong antimicrobial activity against P. aeruginosa (MIC: 5 µg/mL), S. aureus (MIC: 50 µg/mL), MRSA (50 µg/mL), and Candida (MIC: 15.5 µg/mL). In vivo, AG-30/5C accelerated re-epithelialization and angiogenesis in MRSA-infected diabetic mouse and porcine models [37]. AG-30/5C significantly improved the blood flow around the wound compared to the LL-37 control group. Another cationic peptide, AH90, isolated from the skin of frogs, promoted wound healing by stimulating transforming growth factor (TGF-β1) in a full-thickness mouse model. The native AH90 peptide was shown to promote faster wound shrinking in mice compared to a positive control, EGF (epidermal growth factor) and the scrambled version of the peptide, called sAH90 [38].
Tylotoin promotes both wound healing via the induction of TGF-β1 and IL-6 secretion, which are responsible factors for wound healing, and angiogenesis by inducing endothelial cell tube formation. Enhanced cell motility and the proliferation of both keratinocytes and fibroblast in vitro and the formation of granulation tissue at the wounded site in a murine model of full-thickness was shown as well [39]. A small peptide, tiger17 (11-mer), promoted wound healing in vitro and in vivo. It enhanced the migration and proliferation of keratinocytes and significantly accelerated wound closure in a murine full-thickness wound model. Tiger17 induced macrophage recruitment to the inflammation site and via the migration and proliferation of keratinocytes, it induced re-epithelialization and granulation tissue formation. Furthermore, tiger17 stimulated the release of TGF-β1 and IL-6, which are responsible for wound regeneration [40]. A relatively small peptide, WRL3 (18-mer), exhibited high cell selectivity by its ability to specifically eliminate MRSA cells (MIC: 2 μg/mL) grown in a co-culture model via the membrane lysis mechanism and efficiently eradicating biofilms of MRSA. In vivo, WRL3 successfully removed bacteria, stimulated pro-inflammatory cytokine secretion, recruited macrophages, and stimulated angiogenesis and wound closure in an MRSA-infected burn mouse model (Figure 2). WRL3 showed to be more efficient than vancomycin in reducing MRSA infections and to reduce the healing time in burn wounds [41]. Therefore, WRL3 might be used not only for wound healing promotion, but also for treating MRSA-related infections in skin burn wounds.
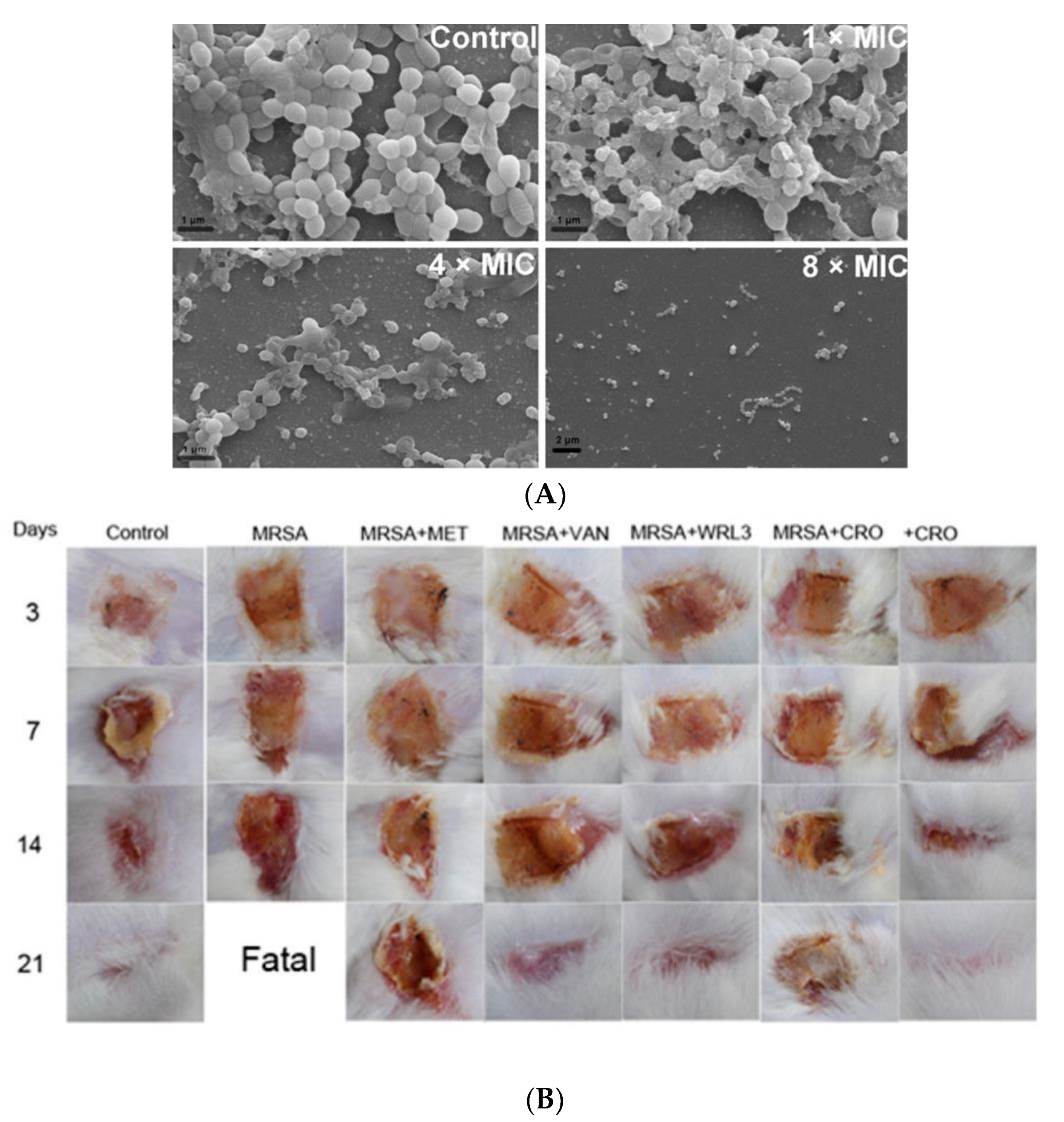
Figure 2. Antibacterial activity of WRL3 and antibiotics. (A) SEM images of the in vitro methicillin-resistant S. aureus (MRSA)-biofilm inhibition at indicated VWR3 concentrations compared to the control cells; (B) Photos of wound regions of approximately 1 cm2 on the back of MRSA-infected mice and treated with MET (methicillin), VAN (vancomycin), WRL3, CRO (ceftriaxone), or WRL3 and CRO at 3, 7, 14, and 21 days post-MRSA infection. “Fatal” indicates no mice survival. From [41] with permission from the American Chemical Society© Copyright 2020.
Another potent AMP, epinecidin-1 (Epi-1), induced keratinocytes proliferation and migration in vitro. In vivo, Epi-1 induced complete healing in an MRSA-infected heat-burned pig skin over a period of 25 days. The results showed that Epi-1 enhanced vascularization, increased epithelial activities, and stimulated collagen synthesis around the wound region [42].
Therefore, AMPs are considered to have a high potential in eradicating microorganisms but as well in promoting wound healing via stimulating re-epithelialization, angiogenesis, collagen, and granulation tissue formation.
The main hurdle of AMPs is their short elimination half-life due to their fast degradation in the presence of proteases once in the blood stream [43]. Therefore, most of the currently used AMPs are directed for topical delivery, especially to wounds. Routinely used AMPs colistin, dalbavancin, daptomycin, oritavancin, and telavancin have a half-life of 5 h, 8 h, 8 h, 14 days, and 195.4 h, respectively, while the half-life of gramicidin has not been determined yet. On average, the median half-life of FDA-approved peptides is 9 h [44]. As the main focus of this review is on the topical application of AMPs, we considered AMPs, which are in their preclinical and clinical trials development stage, as well those routinely used FDA-approved AMPs and potent AMPs for topical wound infections, which possess antibacterial or in specific cases antifungal activity (Table 2).
Table 2. Selected AMPs for topical application under different phases of preclinical and clinical trials, including AMPs FDA-approved and some potent AMPs based on their reported activities.
|
AMPs |
Mechanism of Action |
Activity Against |
Side Effects |
Application and Administration Route |
Ref |
|
Potent AMPs |
|||||
|
AG30 |
Membrane disruption |
Gram (+), (−); E. coli (MIC: 40 µg/mL) and S. aureus (MIC: 20 µg/mL) |
Lack of stability in saline |
Topical |
[36] |
|
AG30/5C |
Membrane disruption |
Gram (+), (−); P. aeruginosa (MIC: 5 µg/mL), S. aureus (MIC: 50 µg/mL) |
More stable than AG30 |
Topical |
[37] |
|
Cys-KR12 |
Membrane disruption |
Gram (+), (−); E. coli (MIC: 4 µg/mL), S. aureus (MIC: 8 µg/mL) |
Not reported |
Topical |
[45] |
|
KR12 |
Membrane disruption |
Gram (+), (−); E. coli (MIC: 2.5 µM), P. aeruginosa (MIC: 10 µM) |
Not reported |
Topical |
[46] |
|
WLBU2 |
Binding to lipopolysaccharide and DNA inhibition |
Gram (+), (−); P. aeruginosa (MIC: 3–8 µM); S. aureus (MIC: 6–16 µM) |
Not reported |
Eradicated lethal P. aeruginosa septicemia in mice |
|
|
WRL3 |
Membrane lysis |
Gram (+), (−); MRSA (MIC: 2 μg/mL) |
Not reported |
MRSA-related infections in skin burn wounds; topical |
[41] |
|
Preclinical Phase |
|||||
|
Arenicin |
Membrane pore formation |
Gram (+), (−); MRSA infection; E. coli (MIC: 1 µg/mL); K. pneumoniae (2 µg/mL) |
Significant toxicity to mammalian cells |
Urinary tract infections; hospital-acquired infections |
|
|
Avidocin and purocin |
Membrane disruption |
Gram (+) and (−) |
Safety reported |
Treatment of C. difficile infections (colitis); topical |
[23] |
|
Buforin II |
Inhibition of DNA/RNA synthesis |
Gram (+), (−); fungi; E. coli (MIC: 1 µg/mL); K. pneumoniae (MIC: 2 µg/mL) |
Safety reported |
Used as bacteriostatic; bactericide; anti-sepsis |
|
|
Lactocin 160 (Bacteriocin) |
Membrane disruption |
G. vaginalis; P. bivia; Lactobacillus spp. (MIC: >200 mg/mL) |
Safety reported |
Urogenital tract infections; Bacterial vaginosis |
|
|
LTX-109 (Lytixar) |
Membrane disruption and cell lysis |
Gram (+); MRSA; 2 VISA; 3 VRSA (MIC: 2–4 µg/mL) |
Itching, pain and burning effects |
Treatment of diabetic foot ulcers; topical |
[23] |
|
Mersacidin |
Inhibition of cell wall |
Gram (+), MRSA (MIC: 2–16 µg/mL); Clostridium spp. (MIC: 1–16 µg/mL); |
Safety reported |
Treatment of hospital-acquired infections |
[54] |
|
Planosporicin (Bacteriocin) |
Inhibition of cell wall |
Gram (+), (−); Planomonospora sp., MDR strains; S. aureus (MIC: 2–>128 μg/mL); S. epidermidis; E. faecalis (MIC: 32 μg/mL) |
Not reported |
Hospital-acquired infections |
|
|
Plectasin (NZ2114) |
Inhibition of cell wall synthesis |
Gram (+), (−); MRSA (MIC: 16–32 μg/mL); P. aeruginosa (MIC >128 μg/mL) |
Not reported |
Pneumococcal peritonitis and pneumonia infections |
|
|
Clinical Phase |
|||||
|
CZEN-002/(CKPV)2 (phase IIb) |
Immunomodulation |
C. albicans |
Not reported |
Vulvo-vaginal candidiasis; topical |
[59] |
|
D2A21 (phase III) |
Membrane disruption |
Gram (+), (−); fungi; Gram (+), (−); S. aureus; E. coli; P. aeruginosa (MIC: 4 µg/mL) |
No side effects reported |
Burn wound infections; topical |
[60] |
|
DPK-060 (phase II) |
Membrane disruption and immunomodulation |
Gram (+), (−); fungi; S. aureus (MIC: 4.6 µg/mL) |
No side effects reported |
Acute external otitis; ear drops |
[23] |
|
EA-230 (phase IIb) |
Immunomodulation |
Gram (−) |
Safety reported |
Sepsis and renal failure protection; IV |
[61] |
|
Histatin (phase I) |
Membrane disruption |
Gram (−); C. albicans (MIC: 4–16 μg/mL) |
No side effects reported |
Treatment of P. aeruginosa infections and oral candidiasis |
|
|
hLF1-11 (phase I/II) |
Membrane disruption |
Gram(+), (−); fungi; Staphylococcus spp. (including MRSA), Streptococcus mitis (MIC: 1.6–6.3 μg/mL); A. baumannii, Pseudomonas spp., Klebsiella spp., E. coli (MIC: 6.3–12.5 μg/mL); Candida spp. (MIC >12.5 μg/mL) |
Little discomfort at the injection site |
LPS-related fungal infections; IV |
|
|
IDR-1 (phase I) |
Immunomodulation |
MRSA; VRSA |
No side effects reported |
Infection prevention in immunocompromised patients |
[62] |
|
IMX942/SGX942 (IDR-1 derivative; phase III) |
Immunomodulation |
Gram (+), (−) |
Safety reported |
Treatment of nosocomial infections, neutropenia |
[62] |
|
LL-37 (phase IIb) |
Barrel-stave mechanism of membrane disruption; inhibit 5LPS binding |
Bacteria, fungi and viral pathogens; P. aeruginosa (MIC: 64 μg/mL) |
Cytotoxic |
Diabetic foot ulcers; chronic middle ear infection |
|
|
LTX-109 (Lytixar, phase II) |
Membrane disruption and cell lysis |
Gram (+); MRSA; VISA; VRSA (MIC: 2–4 µg/mL) |
Itching, pain and burning effects |
Treatment of nasal MRSA infections; nasal and topical |
|
|
Mel4 (phase II/III) |
Membrane disruption |
Gram (+), (−); P. aeruginosa (MIC: 62.5–250 μg/mL) |
No cytotoxicity and no resistance reported; no staining of human cornea |
Contact lenses |
|
|
Melimine (phase I/II) |
Membrane disruption |
Gram (+), (−); P. aeruginosa (MIC: 250–500 μg/mL) |
No cytotoxicity and no resistance reported; staining of human cornea |
Contact lenses |
|
|
MX-226 (Omiganan®; phase III) |
Cell disruption |
Gram (+), (−); MRSA (MIC: 2–8 μg/mL); E. coli (MIC: 8–16 μg/mL); C. albicans (MIC: 64 μg/mL) |
Not reported |
Prevention of device-related infections; topical |
[71] |
|
Novexatin (NP213; phase IIb) |
Membrane disruption |
Fungi |
Not reported |
Treatment of dermatophyte fungal infections |
[57] |
|
Opebacan (rBPI21, neuprex; phase II) |
Membrane disruption |
Gram (+), (−) |
Fever reported |
Meningococcal, wound, and burn infections; IV |
[15] |
|
OP-145 (LL-37 derived; phase II) |
Membrane disruption |
Gram (+) |
Lytic to human cells at high concentrations |
Treatment of chronic bacterial middle ear infection; ear drops |
[72] |
|
PAC113 (P113; histatin 5 analog; phase IIb) |
Membrane disruption and immunomodulation |
Gram (+), (−); Candida spp.; A. baumannii (MIC: 38 μM); P. aeruginosa (MIC: 47 μM), E. cloacae (MIC: 90 μM)
|
No cytotoxicity reported |
Oral candidiasis in HIV patients and prevention of bacterial periodontal disease; topical |
|
|
p2TA (AB103; phase III) |
Immunomodulation |
Gram (−) |
No adverse effects reported |
Necrotizing soft tissue infections; IV |
[74] |
|
Ramoplanin (NTI-851; phase II) |
Membrane disruption and cell wall synthesis |
Gram (+); C. difficile; 4 MSSA (MIC: 2–4 μg/mL) |
Low local tolerability when injected IV |
Treatment of C. difficile-associated infections; oral |
|
|
FDA Approved |
|||||
|
Anidulafungin (Eraxis™) in 2006 |
Inhibition of (1,3)-β-D-glucan synthase |
Fungi; C. albicans and K. crusei (MIC: 0.06 μg/mL) |
Hypersensitivity; hepatic effects |
Treatment of Candida infections, especially esophageal candidiasis; IV infusion |
|
|
Caspofungin (Cancidas) in 2001 |
Inhibition of β (1,3)-D-glucan production |
Fungi; C. albicans; (MIC: 0.06 μg/mL); K. crusei (MIC: 0.25 μg/mL) |
Hypersensitivity; hepatic effects |
Treatment of esophageal candidiasis; IV |
[79] |
|
Dalbavancin (Dalvance™) in 2014 |
Inhibition of bacterial cell wall synthesis |
Gram (+); S. aureus (MIC: ≤0.008–0.5 μg/mL) |
May cause nausea, headache, and diarrhea |
Treatment of complicated skin and skin structure infections (cSSSI); IV injection |
|
|
Daptomycin (Cubicin®) in 2003 |
Membrane lytic |
Gram (+); MRSA (MIC: 0.25–1 µg/mL); VISA (MIC: 1–8 µg/mL); VRSA (MIC: 0.12–1 µg/mL) |
Not approved for pediatric patients |
Treatment of cSSSI; IV injection |
|
|
Gramicidin (Neosporin®) in 1955 |
Pore-forming; aggregation; membrane disruption |
Gram (+), (−); E. faecalis (MIC: 8–16 μg/mL) |
Hemolytic activity |
Treatment of bacterial conjunctivitis; ointment |
|
|
Polymyxins (Polymyxin E = colistin) in 1964 |
Membrane disruption |
Gram (−); P. aeruginosa (MIC: 8 μg/mL); E. coli (0.5 μg/mL) |
Used only as “last-resort” due to neuro- and nephrotoxic effects and neuromuscular blockage |
Skin burns, for washing of wounds after operations, in superficial eye infections, and to protect minor wounds against infections; cream, ear and eye drops |
|
|
Oritavancin (Orbactiv®) in 2014 |
Inhibition of bacterial cell wall synthesis and disruption of bacterial membrane1 |
Gram (+); MRSA and MSSA (MIC: ≤0.008–0.25 μg/mL) |
Long-term treatment is ambiguous |
Treatment of (cSSSI); IV |
|
|
Telavancin (Vibativ ™ and Vibativ®) in 2009 |
Inhibition of bacterial cell wall synthesis and disruption of bacterial membrane1 |
Gram (+); VRSA (MIC: 2–4 μg/mL); VISA (MIC: 0.25–1 μg/mL) |
May induce acute kidney injury |
Treatment of cSSSI; IV |
|
|
Vancomycin (Vancocin® HCl) in 2016 |
Inhibition of bacterial cell wall synthesis |
Gram (+); Candida spp. (MIC: >256 µg/mL); MRSA (MIC: 0.5–2 µg/mL); VISA (MIC: 4–8 µg/mL); VRSA (MIC: 32–>64 µg/mL) |
May cause nephrotoxicity |
Treatment of severe MRSA infections; IV and oral |
|
1Oritavancin and telavancin may also act by membrane-pore, channel formation, or lysis of the cell membrane; 2VISA: vancomycin-intermediate S. aureus; 3VRSA: vancomycin-resistant S. aureus; 4MSSA: methicillin-susceptible Staphylococcus aureus; 5LPS: lipopolysaccharides; Gram (+): Gram-positive bacteria; Gram (−): Gram-negative bacteria. MIC: minimal inhibitory concentration.
Currently, at least 10 AMPs failed in their clinical phases [3]. As examples, pexiganan (Locilex®), which was used for treatment of infections in diabetic foot ulcers, failed during clinical phase III. Iseganan (IB-367, protegrin-I analogue), used for preventing polymicrobial oral infections such as stomatitis, failed in phase III as well. Omiganan (indolicidin derived; MBI-226) failed during phase III due to catheter-related infections [10]. XMP-629 was used to treat impetigo and acne rosacea, and Murepavadin (POL7080) [72], a protegrin analogue used to treat ventilator-associated bacterial pneumonia, was halted recently in phase III. These failures were attributed to the lack of significant efficacy compared to other antibacterial drugs or to multiple side effects [23].
2.1.2. The Challenge of Resistance Development
All types of pathogens tend to develop their own defense mechanism [62], adapting to their specific environment. Therefore, it is unrealistic to expect no development of resistance to AMPs. Generally, microbes create resistance via different modalities, such as modification of their structure or electrical charge [89], modification in the cell wall and metabolism, LPS myristylation, acylation of lipid A, etc. [90]. Additionally, microbes adapt to survive at high ionic concentration, which is a condition known to hinder AMP potency [62].
However, it should be noted that bacteria will develop resistance to AMPs at a much slower rate than to antibiotics [91]. This is attributed to the relatively low specificity of AMP mechanisms of action, as well as to the variety of these mechanisms implied in bacterial killing. In general, AMPs are quite resistant to the development of bacterial resistance [69].
2.1.3. AMP’s Activity against Biofilm Formation
Although AMPs show high activity against a broad spectrum of bacteria, very little is known about the resistance mechanisms developed by biofilms. Biofilm formation by bacteria is another critical issue for infectious diseases. Most pathogens form microcolonies by adhering to each other and then produce a biofilm as a protective environment. Biofilm formation is initiated by (i) planktonic cell attachment to a surface; (ii) colony formation; (iii) biofilm formation, when bacterial colonies are anchored tightly within a matrix of extracellular polymeric substances (EPS), which includes extracellular DNA, proteins, lipids, and polysaccharides; and (iv) cell dispersion [92]. Further, the developed biofilm dispersion may lead to the spread of infection and new colonization sites and may create increased antimicrobial resistance.
Severe infections are associated with biomedical devices and implants due to their high susceptibility to bacterial colonization [93]. Very often, implant surfaces become a reservoir of bacteria that can spread very fast into the whole body, which leads to persistent chronic infections. The only way to eradicate bacteria in this situation is implant removal, which implies surgery and close follow-up of the patients for their complete recovery. In this context, it is worth noticing that AMPs, such as AG-30 [36], AG-30/5C [37], WRL3 [41], melimine and Mel4 [69], 73c, and D-73 [94] are very potent against biofilm formation.
2.2. Antimicrobial Dendrimer Peptides
Antimicrobial peptide dendrimers (AMPDs) are highly branched 3D structures with a central core and high density of flexible surface groups for potential molecule attachment [95]. They can be obtained by solid-phase peptide synthesis (SPPS) and purified by HPLC to ensure high reproducibility [96]. Compared to polymeric dendrimers, they are built on covalently coupled amino acid residues [92]. AMPDs can be built up to several generations based on the layers of branching units from G0 to Gx, where x represents the number of branching cycles (Figure 3). They are a very attractive class of molecules as they exhibit high antimicrobial activity against bacteria, fungi, and viruses [97]. Their high antimicrobial activity is mainly due to their positive charges of the amino acid residues that interact with the negatively charged bacterial cell envelope and lead to bacterial death [92][98]. Moreover, AMPDs were shown to be more resistant to proteolysis and less toxic to both mammalian and erythrocyte cells than AMPs.
In addition to their intrinsic activity and due to the fact that AMPDs have different functional groups, it makes them very attractive delivery systems as drug carriers. Increasing the number of ramification leads to an increase in functional groups, thus a higher opportunity to couple different bioactive molecules [95].
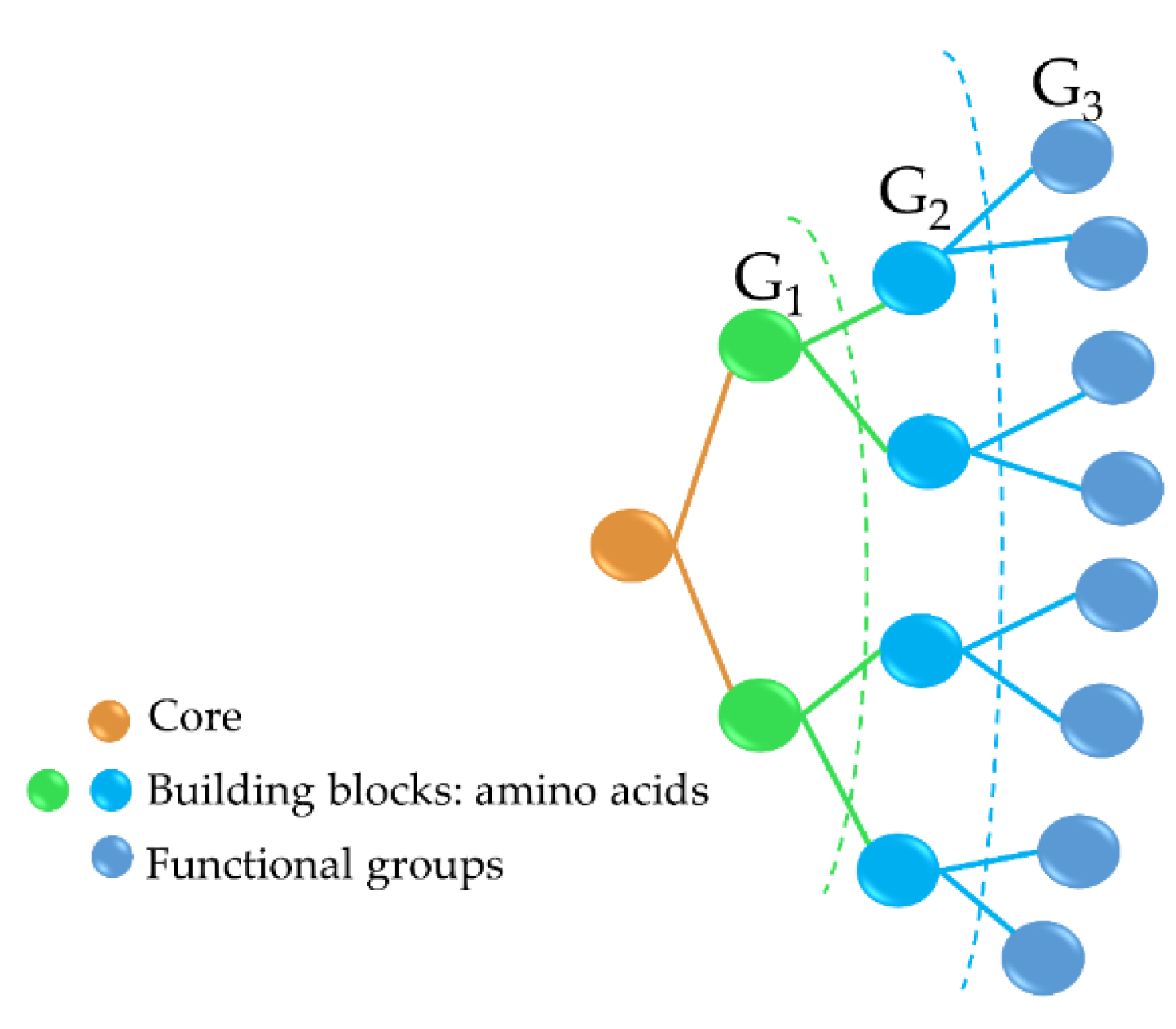
Figure 3. Third-generation AMPD structure, including the dendrimer core (peptidic or non-peptidic), building blocks (amino acids), and functional groups (peptidic or non-peptidic) for coupling other bioactive molecules. G1, G2, and G3 indicate the different generation levels.
Bacterial killing by AMPDs is governed by functional groups of the amino acid residues and their ability to penetrate the cell membrane [92]. A recent example of effective third generation (G3) AMPD, G3KL (incorporating repetitive lysine (K) and leucine (L) units), showed high activity against 4 out of 6 in the ESKAPE collection in 32 different strains of A. baumannii (MIC: 16 µg/mL), 35 strains of P. aeruginosa (MIC: 8–32 µg/mL) [99], E. coli (8 µg/mL), and K. pneumoniae (16–64 µg/mL) [96]. The mechanism of action is based on membrane disruption followed by vesicle leakage as shown by transmission electron microscopy (TEM) analysis. G3KL AMPD showed as well to inhibit P. aeruginosa biofilm formation [100]. Fluorescently labeled G3KL entirely diffused into P. aeruginosa within 15 min as shown in a time-lapse assay, suggesting AMPD diffusion or translocation into the cytoplasm through damaging both the outer and inner bacterial membrane, as was confirmed by TEM [101].
G3KL and another G3 (G3RL, consisting of arginine (R) and leucine (L) repeats) showed high antibacterial activity against P. aeruginosa and neither toxicity nor gene alteration toward progenitor fibroblasts at an elevated concentration of 100 µg/mL. Furthermore, G3KL better promoted angiogenesis than G3RL in a human umbilical vein endothelial cells (HUVEC) and chorioallantoic membrane (CAM) model (Figure 4) [5], which is an important aspect for accelerated wound healing.

Figure 4. Images of the endothelial tubular networks in Matrigel after 5 h treatment with G3KL (incorporating repetitive lysine (K) and leucine (L) units), and G3RL (consisting of arginine (R) and leucine (L) repeats) at indicated concentrations versus PBS (Phosphate-Buffered saline) control. From [5] with the permission by Springer Nature©, Copyright 2016.
A second generation (G2) AMPD, TNS18, showed the same activity as G3KL against P. aeruginosa (MIC: 8–16 µg/mL), a much higher activity against S. aureus (MIC: 8–16 µg/mL), but less potency against K. pneumoniae than G3KL [96]. Combination of the peripheral branches of G2 (TNS18) with the core of a G3 (called T7) resulted in a chimeric AMPD named DC5. DC5 displayed significant activity against K. pneumoniae (MIC: 16–32 µg/mL), MRSA (MIC: 32 µg/mL), E coli (MIC: 8 µg/mL), A. baumannii (MIC: 16 µg/mL), and P. aeruginosa (MIC: 4–8 µg/mL) strains [96]. In vivo on larvae, D-enantiomers of dG3KL and dTNS18 showed a high ability to kill P. aeruginosa biofilm (killing efficiency 90.2–100%) and similar to tobramycin [102].
Another family of AMPDs based on R4 tetrapeptide (RLYR repetitive units) and R8 octapeptide (RLYR-KVYG repeats) was tested against 10 different microbial strains (E. coli, P. aeruginosa, P. vulgaris, K. oxytoca, S. aureus, M. luteus, E. faecalis, C. albicans, C. kefyr, and C. tropicalis) with lower MICs than 1 µM. Noteworthy, they proved as well to be more resistant to proteolysis and more non-hemolytic than the corresponding linearly repeating peptides [97]. A SB056 lipodimeric AMPD exhibited high microbicidal activity against both Gram (+) and (−) bacteria with an MIC as low as 2–32 µg/mL against A. baumannii, E. cloacae, E. coli, K. pneumoniae, and P. aeruginosa, which is comparable activity to polymyxin B. Importantly, SB056 strongly inhibited E. coli, S. aureus, and S. epidermis biofilm formation [103].
These AMPDs with preferred selectivity have proven to be highly active against the whole range of ESKAPE bacterial pathogens and others, including fungi as well. Additionally, SB105-A10 AMPD exhibits high antiviral activity against human papillomaviruses, human respiratory syncytial virus (RSV), and human immunodeficiency virus type 1 (HIV-1), as described elsewhere [95]. Therefore, further studies are warranted to bring good news in the fight against AMR in infection-related diseases. So far, there are no further studies with regard to the coupling of AMPDs with AMPs or other chemistry involved to deliver AMPDs to the wound site.
3. Peptide Conjugates
Needless to mention, most of the recent studies report on new strategies to combat MDR and biofilm formation. It is clear now that the antimicrobial activity could be considerably enhanced by several strategies, such as the chemical addition of different molecules via covalent grafting, incorporation into different nanosystems, self-assembly, coupling with antibiotics, and incorporation into polymer scaffolds, while maintaining or improving AMP’s antibacterial activity with prolonged blood circulation and reduced cytotoxicity. Other approaches for “tailoring” AMPs with enhanced stability, such as modification of the C- and N-terminus via acetylation and amidation, cyclisation, lactamization, lactonization, macromolecular conjugation, and PEGylation have been discussed elsewhere in detail [62][104][105].
Another potential strategy to improve the efficacy of current antibacterial agents is to use antibiotic resistance breakers (ARBs). ARBs are a type of antibiotic adjuvants that are capable of re-sensitizing resistant bacteria to antibiotics. The three main classes of ARBs include β-lactam inhibitors, membrane permeabilizers, and efflux pump inhibitors (EPIs) [106][107]. Among ARB–antibiotic combinations, the co-administration of β-lactamase inhibitors (e.g., clavulanic acid) with β-lactam antibiotics (e.g., amoxicillin) has met clinical success and been extensively described in the literature [106][107][108].
3.1. Covalent Coupling to Polymers
Polymers are composed of several repeating units of monomers. Many natural polymers, such as proteins (i.e., collagen, elastin, keratin, and silk fibroin) and polysaccharides (i.e., alginate, chitosan, hyaluronic acid, and cellulose), or synthetic polymers (i.e., poly(lactic acid) (PLA), poly(vinyl alcohol) (PVAL), poly(caprolactone) (PCL), poly(lactic-co-glycolic acid) (PLGA), and poly(ethylene glycol (PEG) have been widely used for medical applications [109]. Natural biopolymers (e.g., chitosan, collagen, and hyaluronic acid (HA)) have been shown to promote wound healing, for instance by stimulating anti-inflammatory responses in chronic wounds [3]. A loss of antimicrobial activity or low availability at the implant site when antimicrobial agents are covalently coupled to different surfaces is a known problem related to antimicrobial coatings [110]. To overcome these issues, AMPs are promising agents with high bioactivity and biocompatibility, which showed relatively low bacterial resistance.
Among biopolymers, chitosan is widely used thanks to its amino and hydroxyl functional groups [111][112], which offer a variety of possibilities for covalent or non-covalent coupling. Chitosan is a biocompatible, biodegradable polymer with antimicrobial activity and the ability to promote wound healing, dermal cell proliferation, and migration [111][112]. Several chitosan–AMP conjugates have been designed, such as Dhvar-5 AMP immobilized to chitosan via azide-alkyne or “click” reaction, which showed promise as antibacterial coatings to prevent biomaterial-related infections. An ultrathin layer was obtained by spin coating when dissolving AMP–chitosan in acetic acid. Higher antibacterial (MIC: 4, 2, 16, and 4 µg/mL for S. aureus, S. epidermidis, E. coli, and P. aeruginosa, respectively) and antibiofilm activities were found when coupling Dhvar-5 peptide via C-terminus than when coupling through the N-terminus, underlining the importance of the conformation of the exposed peptide. Moreover, the AMP–chitosan conjugate did not show any cytotoxicity to fibroblasts [113]. Similarly, hLF1-11 AMP was covalently immobilized by C-terminal onto chitosan thin films; the presence of hLF1-11 inhibited the growth of MRSA in comparison to the control [64]. In another study, cysteine-modified MSI-78 AMP was covalently immobilized to chitosan coatings through a PEG linker. MSI-78–chitosan films showed high antimicrobial efficacy against S. epidermidis in both PBS and PBS supplemented with 1% human plasma, maintaining significantly low bacterial adhesion, while killing 76% of adherent bacteria [114]. This type of coating could be successfully used for medical applications for implants or wound dressings.
PEG has been extensively used to functionalize peptides, drugs, and nanoparticles. This process of PEGylation offers different advantages, such as increased resistance to degradation, increased blood circulation time, reduced aggregation, and reduced toxicity [115]. Sometimes, peptide PEGylation may lead to reduced proteolysis and toxicity at a cost of lower antibacterial effect. As such, Imura et al. PEGylated tachyplesin I and found lower antimicrobial activity than the non-PEGylated peptide against E. coli and significantly lower toxicity against CHO-K1 cells [116]. In another study, the same authors loaded unilamellar vesicles with PEGylated magainin 2 and found that PEG-magainin 2 had weaker vesicle leakage, lower bactericidal activity, and significantly lower toxicity to CHO-K1 cells [117]. To investigate this decrease in antibacterial activity, Singh et al. PEGylated the KYE28 AMP using PEG with different lengths and conjugation sites. They found that the antibacterial activity decreased against E. coli and P. aeruginosa and dramatically decreased toward S. aureus with the increase of PEG length, irrespective of the conjugation site. Cytotoxicity and hemolysis were strongly reduced as well, while selectivity was improved (Figure 5) [118]. Thus, PEGylation is an attractive technique to optimize the antibacterial efficacy of the peptide versus their toxicity. In another study, peptide 73, a derivative of aurein 2.2Δ3, coupled to PEG exhibited a 2- to 8-fold enhancement of antibacterial activity against S. aureus biofilms [94].
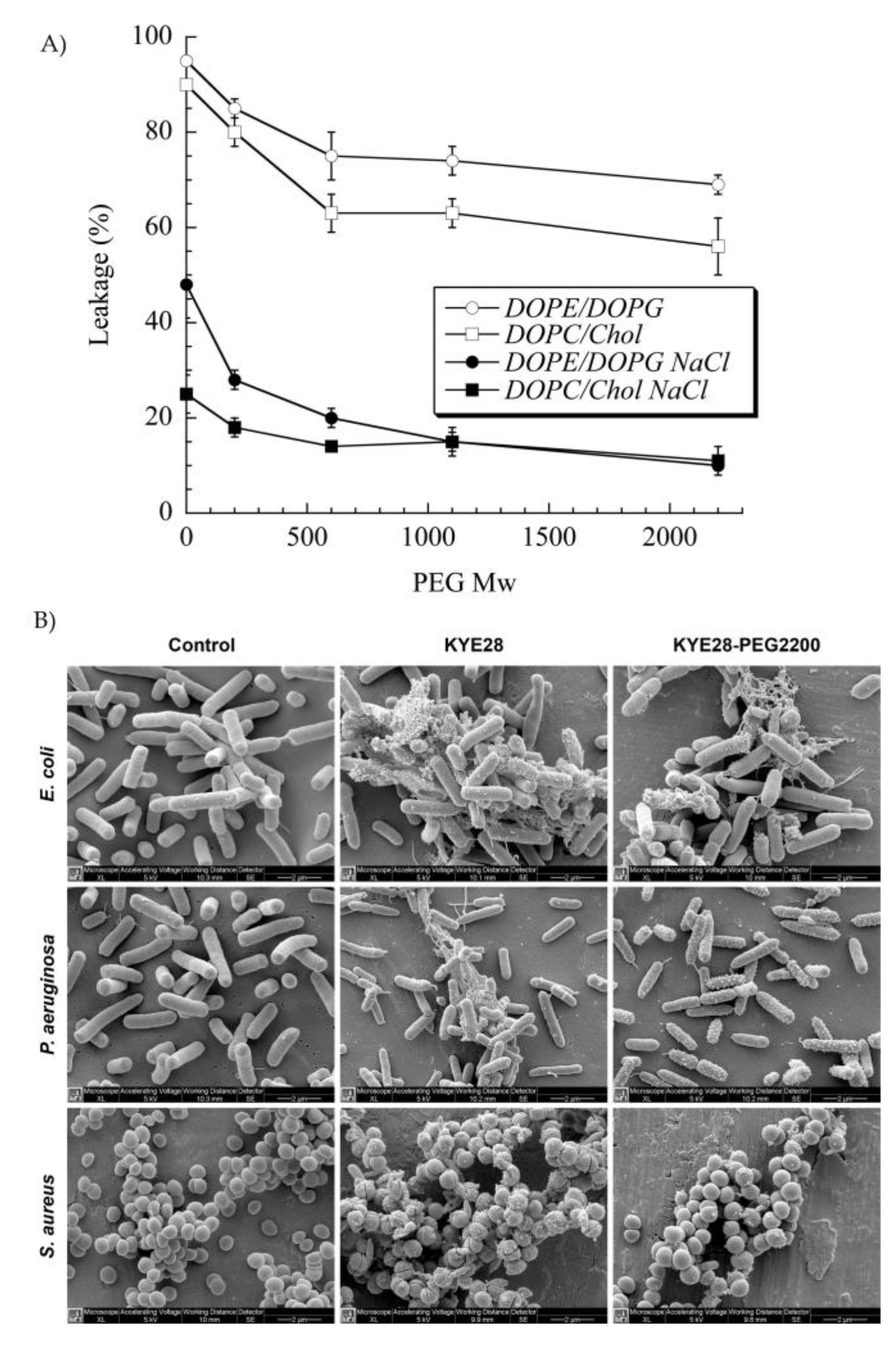
Figure 5. (A) Effect of poly(ethylene glycol (PEG) length on KYE28–PEG leakage induction of DOPE/DOPG (75/25 mol/mol) and DOPC/cholesterol (60/40 mol/mol) liposomes at a peptide concentration of 1 μM. (B) Peptide-mediated permeabilization of E. coli, P. aeruginosa, and S. aureus with the indicated peptides at 30 μM for 2 h, and analyzed by SEM. DOPE: 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine, DOPC: 1,2-dioleoyl-sn-glycero-3-phosphocholine, and DOPG: 1,2-dioleoyl-sn-glycero-3-phosphoglycerol, monosodium salt. Reprinted from [118] with the permission of American Chemical Society©, Copyright 2014.
In general, the bacterial colonization of medical devices favors biofilm development, which in turn will lead to device failure due to peri-implantitis [119]. For instance, P. aeruginosa colonization of catheters and other medical devices accounts for 10–20% of all related infections [69]. Therefore, coating medical devices with a layer of antibacterial membrane or covalently attaching different AMPs to surfaces are advantageous strategies to combat biofilm formation and reduce the rate of infection. In this view, two cationic AMPs, melimine and Mel4 (melimine derivative), which are in phase II/III clinical trials, were covalently attached to a model glass surface [69]. Surfaces coated with both peptides lead to cytoplasmic bacteria leakage within 15 min. Moreover, the antimicrobial activity of melimine was preserved upon covalent coupling to polymers or titanium, and melimine-coated lenses did not show signs of conjunctival redness, although staining of human cornea was observed in some cases [120]. On the contrary, Mel4-coated lenses showed no signs of corneal staining or redness or ocular irritation [121]. Moreover, covalently coupled melimine and Mel4 to the glass surface was shown to successfully eradicate P. aeruginosa within 15 min [69]. Overall, AMP–polymer conjugates can confer effective antibacterial properties to the surfaces of medical devices at the preclinical stage.
3.2. Self-Assembled AMPs
Peptides can self-assemble into different nanostructures, such as micelles, nanotubes, vesicles, and fibrils. Their self-assembly mechanism is driven by the spontaneous organization of peptides based on their electrostatic or hydrophobic interactions, hydrogen bonding, or π–π stacking [122]. An important focus of self-assembled peptides research relates to the formation of amyloids, which is a key process for the onset of neurodegenerative diseases, especially with regard to the link between microbial infection and Alzheimer’s, Parkinson’s, Creutzfeldt–Jakob, or related diseases [123][124][125][126]. For example, Alzheimer’s β-amyloid diphenylalanine (FF) dipeptide was found to drive the self-assembly of FF into stiff nanotubes in Alzheimer`s disease [127]. A β-amyloid KLVFF peptide that has the same mechanism as FF peptides could self-assemble into nanofibrils and then into a nanofibrillar gel in a concentrated phosphate-buffered saline solution [128]. Additionally, amyloid peptides such as islet amyloid polypeptide (IAPP) exhibited antimicrobial activity, and sometimes, their activity was higher than that of LL-37 [129]. Therefore, AMPs could suggest the potential connection between amyloid and AMPs in combating amyloid-related neurodegenerative diseases. AMP may be of therapeutic interest in the treatment of neurodegenerative diseases, since persistent microbial infection is thought to be one of the triggering factors of these diseases.
Melittin, a very potent AMP extracted from bee venom but severely cytotoxic, has been co-assembled upon the addition of a molecular block containing synthetic multidomain peptides. Upon the self-assembly of melittin into nanofibers, membrane selectivity and cytocompatibility was significantly improved [130]. Yazici et al. combined a hydroxyapatite-binding peptide-1 (HABP1) with a tet127 AMP through a flexible linker as self-assembled antimicrobial agents against E. coli (MIC: 32 µg/mL) and less efficiently against Streptococcus mutans (MIC > 256 µg/mL). When using these self-assembled HABP1-tet127 to functionalize calcium–phosphate coated on nanotubular titanium surfaces, a better inhibition of E. coli (90%) than S. mutans (75%) was shown [131]. This type of self-assembled peptides could be used for functionalizing the surfaces of different medical implants for local therapy to prevent bacterial infection. Bacitracin A modified with poly(D,L-lactic-co-glycolic acid) (PLGA) and poly(ethylene glycol) (PEG) exhibited self-assembling properties with high antibacterial potency both in vitro and in vivo. All PEGylated self-assemblies showed significantly stronger antibacterial activity against Gram (+) and Gram (−) compared to the non-PEGylated system. The antibacterial activity for non-PEGylated self-assemblies was severely compromised due to low water solubility. In contrast, the PEGylation of bacitracin–PLGA self-assemblies significantly improved water solubility of the system and in turn, the antibacterial activity was improved against E. coli (MIC: 1 µM) and S. aureus (MIC: 0.5 µM) without significant toxicity in a mouse thigh infection model. Bacitracin in solution, used as control, was effective only against Gram (+) bacteria with MICs between 2 and 4 µM, while MICs against Gram (−) were higher than 128 µM. It was shown as well that PEGylation did not affect antibacterial activity; on the contrary, PEGylated bacitracin-PLGA exhibited high accumulation in inflammatory tissue and prolonged blood circulation [132]. This system could be efficiently used to design novel antibiotic nano-assemblies in the treatment of invasive infections.
Self-assembly is a promising strategy toward the design of smart materials that could incorporate antibiotics or other antimicrobial agents with stimuli-responsive properties. For example, pH-sensitive materials could release antibiotics at a pre-defined pH, ionic strength or temperature, enhancing activity in the targeted infected wounds. Moreover, due to their high biocompatibility, biodegradability, and membrane selectivity, AMP self-assemblies hold great potential toward eradicating MDR and biofilm-forming bacteria. Self-assembled peptides and factors influencing AMP self-assembly have been described by Malekkhaiat Häffner and Malmsten in detail [126].
3.3. Combination of AMPs with Antibiotics
AMPs associated with conventional antibiotics generally showed high synergism in terms of their antimicrobial activity [62]. This is attributed to the fact that the combined antibiotic/antimicrobial therapy causes bacterial cell membrane disruption, facilitating antibiotic penetration and accumulation in the cytoplasm, ultimately leading to cell death. For example, magainin 2 and cecropin A combined with rifampicin showed synergistic interactions by significantly inhibiting MDR P. aeruginosa development in vitro and in vivo in rats. The best in vivo results on mortality rates and bacteraemia were obtained when administering intraperitoneally the combination of rifampicin (10 mg/kg) with either magainin 2 (dose of 1 mg/kg) or cecropin A (1 mg/kg). Mice treated with rifampicin (10 mg/kg), magainin 2 (1 mg/kg), or cecropin A (1 mg/kg) alone showed a high mortality rate and high bacterial survival, respectively [133]. This finding suggested that the membrane-permeabilizing activity of the peptides allows rifampicin to gain access to its intracellular target. Stronger synergistic effects have been observed in vivo in a mouse model of sepsis than in vitro when combining tachyplesin III with imipenem [134]. Synergistic effects against MDR P. aeruginosa were seen when combining a peptide (18 mer) called “P5” with isepamicin. Isepamicin could enter bacterial cells and inhibit protein synthesis assisted by P5, which lysed the cell membrane [135]. Such synergy was not observed upon mixing P5 with cefpiramide. Thus, combinations of P5 with isepamicin may be used for the treatment of patients with cholithiasis, which is a condition affected by antibiotic-resistant bacteria.
Similarly, B2088 AMP in combination with different conventional antibiotics, such as chloramphenicol, tobramycin, gentamicin, and imipenem exhibit synergistic antibacterial effects against P. aeruginosa without cytotoxicity against mammalian cells [136]. For instance, the WRL3 peptide combined with ceftriaxone exhibited synergistic effects against MRSA-infected burn wounds in mice [41]. This AMP–antibiotic mixture could be efficiently used in clinical applications for burn wound infections, where MRSA and P. aeruginosa cause high rates of death.
Therefore, the combined administration of AMPs and antibiotics appears to be a promising strategy and is already applied in clinics for its synergistic antibacterial effects and ability to fight MDR bacteria causing infectious diseases. Additionally, AMPs/antibiotics synergies against difficult-to-eradicate biofilms have been reported [137][138][139].
4. Nanotechnological Platforms and Scaffolds for Peptide Delivery
As mentioned before, stability, short half-life, and cytotoxicity are the key factors limiting most of the AMPs from further clinical application. During the last few decades, a plethora of studies have been performed to find different strategies or systems to deliver AMPs. Nanotechnology is the field of science that uses nanocarriers such as nanoparticles (NPs) as drug delivery systems. NPs are particles in the range of 0.1 to 100 nm. The conjugation of AMPs with NPs results in increased local concentration at the delivery site with an enhanced AMP bioactivity, which might be attributed to a synergistic effect [140]. Nanotechnology can add many advantages, such as the improvement of solubility, bioavailability, release, and higher penetration within biofilms of AMPs [141][142]. They can provide protection to AMPs against degradation in different environments, such as enzymatic degradation [35].
4.1. Polymeric Scaffolds and Wound Dressings
Polymer scaffolds are the most used formulations to address wound healing and recovery due to their ability to promote cell attachment and proliferation, extracellular matrix generation, and the restoration of vessels via creating physical bonds between the cells and 3D network of the scaffolds [143]. Moreover, scaffolds can retain a significant amount of water or biological fluids within their 3D network due to the interaction of water with polymer hydrophilic groups (hydroxyl, amine, amide, and carboxyl groups), or hydrophobic interactions with specific biological fluid components and/or osmotic driving force effects [144]. Polymer-based scaffolds can be applied at the wound site and be used as reservoirs for the delivery of AMPs via diffusion, swelling, or chemical degradation [3][145].
In this view, a new strategy for topical application was developed by tethering human cathelicidin LL-37 on collagen scaffolds for the treatment of wound infections. LL-37-loaded scaffolds exhibited no toxicity toward fibroblast at a peptide concentration 24-fold higher than the cytotoxic threshold [146]. After 14 days, LL-37 loaded onto collagen domains (derived from collagenase or fibronectin) was retained on the scaffolds and showed no inhibition of antimicrobial activity against both Gram (+) and (−). This poor delivery may be due to too strong peptide–scaffold interactions. Similarly, Cassin et al. evaluated the incorporation of LL-37 into collagen and hyaluronic acid polyelectrolyte multilayers (PEMs) via physisorption and covalent immobilization [147]. LL-37-loaded thin films effectively prevented E. coli adhesion, but cells treated with covalently immobilized AMP presented morphological changes, suggesting cytotoxicity. In another study, Cys-KR12 AMP, a LL-37 derivative, was covalently immobilized on electrospun silk fibroin (SF) nanofiber membranes via EDC/NHS (1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide/ N-hydroxysuccinimide) and thiol-maleimide “click chemistry”. Chemical immobilization of the AMP to SF membranes did not inhibit the biological activity of the peptide. On the contrary, the conjugate was highly active against four different strains, such as S. aureus, S. epidermidis, E. coli, and P. aeruginosa, with no biofilm formation (Figure 6) [45]. Cys-KR12-SF membranes promoted the proliferation of keratinocytes and fibroblasts, and they induced the differentiation of keratinocytes with a pronounced cell adhesion, which are important key steps in wound healing. Cys-KR12 (20 μg/mL) was used as positive control. These membranes are promising candidates for topical wound-healing applications.
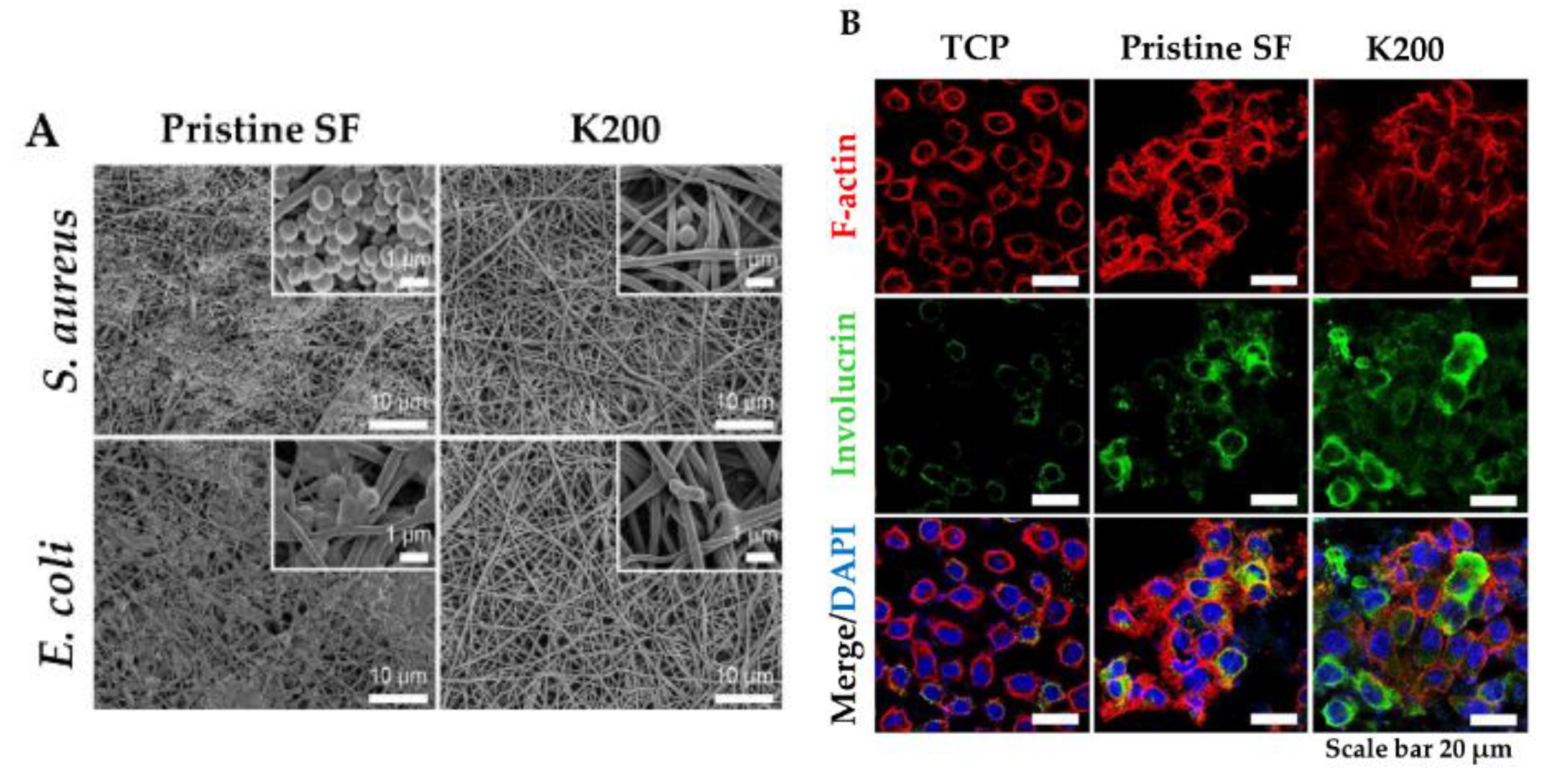
Figure 6. (A) FE-SEM images of S. aureus and E. coli cultured on Pristine SF and Cys-KR12 at 200 µg/mL (K200). (B) Confocal immunofluorescence images of keratinocytes cultured on TCP (tissue culture treated plates), pristine SF and K200 (red channel, F-actin; green, involucrin; blue, DAPI (4′,6-diamidino-2-phenylindole)). Reprinted from [45] with permission from Acta Materialia Inc.©, Copyright 2016.
Cellulose and its derivatives loaded with AMPs have been tested in diabetic foot ulcer (DFU) treatment. In a randomized phase III clinical trial [148], ACT1 AMP embedded in a hydroxyethyl–cellulose (HEC) hydrogel (Granexin) was topically applied for DFU treatment. The results showed that Granexin treatment was safe and effective for DFU treatment with a significant reduction of the ulcer area at week 12 (72% reduction versus control with 57%). All patients treated with ACT1-based hydrogels reached 100% ulcer re-epithelialization in a short time with no toxicity, no immunogenicity, or any side effects reported [149]. Such systems confirm their utility for the treatment of topical wound infections, especially DFU patients.
All these studies support the potential for AMP loaded into polymer scaffolds for the sustained and enhanced biological activity of the AMPs. Designing a polymer-based scaffold loaded with AMP that could control the AMP release via binding to the matrix, while promoting wound healing via the bioactivity of the scaffold itself, would be beneficial not only for wound healing, but also for preventing bacterial infection.
4.2. Organic Nanoparticles
4.2.1. Polymeric Nanoparticles, Nanoemulsions, and Micelles
Polymer nanoparticles may be produced either in the form of gel-like particles or solid polymer particles. Nano- or microgel particles are prepared from swollen hydrophilic polymers; they are generally obtained by ionotropic gelation, a one-step process in which a counter-ion is added to a polyelectrolytic polymer, thus producing micro- or nanoparticles though ionic interactions. In contrast, solid polymer particles are based on organo-soluble, poorly hydrophilic polymers such as PLGA or PLA, and they may be produced by methods such as oil-in-water or water-in-oil-in-water emulsion evaporation.
Carboxymethyl chitosan (CMC) NPs have been loaded with a very potent AMP OH30 extracted from king cobra for potential wound healing. Surprisingly, positively charged CMC could interact with negatively charged bacterial cell membranes and assist the internalization of the OH30 peptide. A slow release of OH30 from the CMC-OH30 NPs was seen over 24 h and maintaining antimicrobial activity as well. CMC-OH30 NPs could slightly enhance migration (95%) but not the proliferation of keratinocytes compared to OH30 (85%), CMC NPs (75%), or untreated cells (60%). In vivo, CMC-OH30 NPs significantly accelerated wound healing in a full-thickness excision mouse model. Moreover, mice treated with CMC-OH30 NPs exhibited 70% wound closure at day 5 compared to CMC NPs or OH30 alone, which maintained wound closure in the range of 36–58% [150]. A drawback of the CMC-OH30 complex is its limited stability as shown by the variability in size and zeta potential over 28 days. Another 5-amino acid AMP, RBRBR, was encapsulated into chitosan NPs through the ionotropic gelation process with 51% encapsulation efficiency, slowly releasing the AMP over 14 days. AMP-loaded chitosan NPs showed at least 3-log increased antimicrobial activity against S. aureus while decreasing the toxicity against both mammalian and human erythrocyte cells. Importantly, positively charged (+33 mV) AMP–chitosan NPs (121 nm) significantly inhibited the growth of biofilm (up to 98%) [151]. The straightforward approach of ionotropic gelation may be used to deliver other potent AMPs while reducing their cytotoxicity, in order to achieve efficient antibacterial effects against MDR and biofilm-forming bacteria.
Nordström et al. investigated the ability of poly(ethyl acrylate-co-methacrylic acid) (MAA) microgels to deliver two AMPs, LL-37 and DPK-060. AMP loading into the microgels decreased toxicity toward erythrocytes and was suggested to protect peptides against protease degradation. In turn, microgel-loaded peptides could kill MRSA, E. coli, and P. aeruginosa via a membrane disruption mechanism [152]. Further, in an effort to elucidate the mechanism of interaction between microgel-loaded AMPs and cellular membranes, Nordström et al. included LL-37 into MAA microgels. The insertion of the peptides released from LL-37-loaded microgels was shown to occur without membrane defect formation at low concentrations, but with the loss of lipid from the bilayer in a concentration-dependent manner [153]. Similarly, LL-37 has been encapsulated into liquid crystalline nanoparticles (LCNPs), such as cubosomes, for the treatment of S. aureus skin infection. LL-37 loaded into cubosomes was protected against proteolytic degradation by P. aeruginosa elastase while improving AMP bioavailability and efficiency. In an ex vivo wound infection model, LL-37-loaded cubosomes showed the highest killing efficiency of S. aureus and an absence of pig skin irritation [154]. Further molecular dynamic simulations confirmed these experimental results, revealing that charged amine and guanidium groups from LL-37 are responsible for facilitating the interaction with the bacterial membrane. Moreover, LL-37 stabilized the cubosome through hydrophobic interactions, while polar residues remained in solution [155].
Silva et al. loaded LLKKK18 (KEFKRIVKRIKKFLRKLV) into hyaluronic acid nanogels and reported a higher stability of the peptide and reduced toxicity to mammalian cells. Importantly, it was found that nanogels were internalized by macrophages infected with Mycobacterium tuberculosis or M. avium. A significant reduction of bacterial load in infected mice after tracheal administration of the nanogels was seen [156]. Similarly, tet213 AMP was admixed to alginate, hyaluronic acid, and collagen for a potential wound dressing. Tet213 dressings showed antimicrobial activity against three different bacterial strains (E. coli, MRSA, and S. aureus) and promoted fibroblast proliferation in vitro. Moreover, in vivo Tet213 dressings promoted re-epithelialization, enhanced collagen deposition, angiogenesis, and the completion of wound healing in E. coli- and S. aureus-infected full-thickness wounds in rats. The AMP dressing had higher antibacterial activity than silver-containing commercial Aquacel, alginate, hyaluronic acid, and collagen dressing [157].
As for solid micro/nanoparticles, LL-37-loaded PLGA NPs were prepared via a water-in-oil-in water (W/O/W) emulsion–solvent evaporation method and showed high antibacterial activity in vitro against E. coli. Topically applied PLGA-LL-37 NPs enhanced wound closure in mouse full-thickness excisional wounds with an average healing of 79% and 90% at days 7 and 10, respectively (Figure 7). Moreover, advanced granulation tissue formation, re-epithelialization, and accelerated angiogenesis was observed in the group of mice treated with PLGA-LL-37 NPs [158]. The only disadvantage of such system is the limited residence time and bioadhesion in the wounds, which is not desired in the case of hard-to-heal wounds. Similarly, esculin1-a AMP loaded into PLGA NPs resulted in selective bioactivity with high antibacterial activity against P. aeruginosa in both in vitro and in vivo and decreased cytotoxicity. Despite that AMP loaded NPs showed lower antibacterial activity in vitro than free esculin1-a at 24 h, they showed a constant inhibition of P. aeruginosa growth up to 72 h. Furthermore, AMP-loaded NPs (0.1 mg/kg) in a mouse model of acute P. aeruginosa lung infection were shown to significantly reduce the bacterial load by 17-fold compared to free esculin1-a in solution [159].
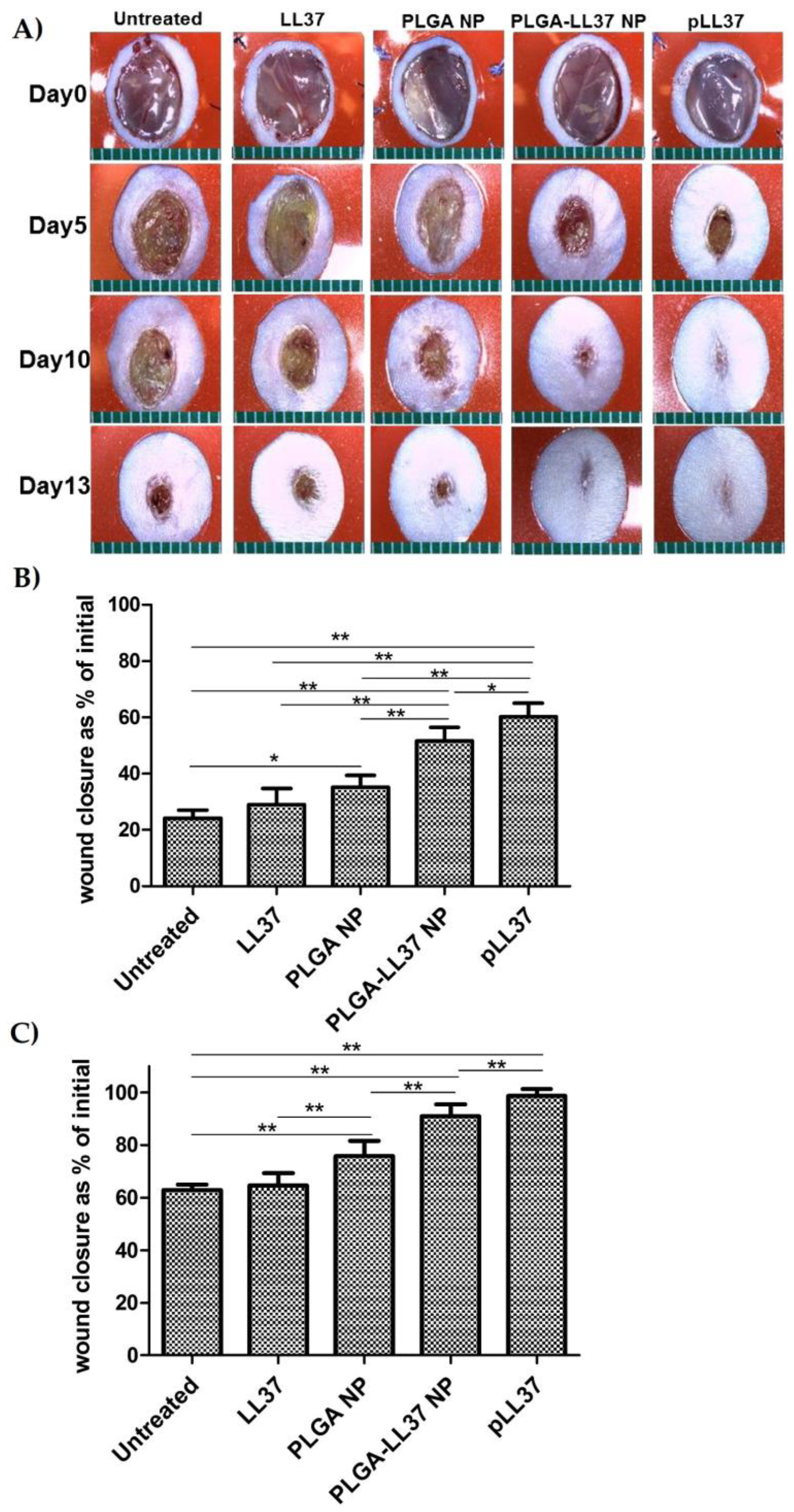
Figure 7. Accelerated wound healing in mice treated with PLGA-LL-37 nanoparticles (NPs) compared to controls. (A) Images of wounds in mice of five tested groups: untreated, LL-37, PLGA-NP, PLGA-LL-37 NPs, and pLL-37 (plasmid encoding hCAP18/LL-37). Representation of wound area at (B) day 5 (n = 13) and (C) day 10 (n = 10) (mean ± SD). Reprinted from [158] with the permission of Elsevier©, Copyright 2014.
The performance of AMP incorporated into micelles was investigated by Wang et al. After complexation process between MSI-78 (pexiganan) peptide and methoxy poly(ethylene glycol)-b-poly(α-glutamic acid) (mPEG-b-PGlu), it was found that hemolytic toxicity was decreased through polyelectrolyte complexation without hindering its antimicrobial activity against E. coli, B. subtilis, and S. aureus [160]. In another study, peptide 73c and D-73 encapsulated into PEGylated micelles showed increased antibacterial activity against S. aureus biofilms by 510- and 9-fold, respectively, compared to their parent peptide in a murine cutaneous abscess model. In vitro, peptides formulated as PEGylated micelles exhibited decreased toxicity to mammalian cells [94].
LL-37-loaded nanostructured lipid carriers (NLC) were prepared by the melt-emulsification method and showed a preserved bioactivity against E. coli (killing efficiency 73%). Topically applied NLC-LL-37 to the wounds enhanced wound closure, re-epithelization, collagen deposition, and angiogenesis in diabetic mice [161]. The major drawback of such a system is the limited residence time in the wounds.
4.2.2. Liposomes
Liposomes as drug carriers have various advantages for providing sustained and controlled drug release for topical applications [162]. Examples of commercially available liposome-based antibiotics that encapsulate amphotericin antibiotic include AmBisome®, Fungisome®, and Amphotec® and lipid-based Abelcet® [163].
In this view, vancomycin was used as a model drug by Yang et al. [164]. Drug-loaded liposomes were coated with chitosan in order to improve vancomycin pharmacokinetics and exhibited sustained drug release both in vitro and in vivo, without inducing hemolysis. Importantly, chitosan-coated vancomycin-loaded liposomes had low concentration in kidneys and high concentration in lungs compared to free vancomycin or vancomycin-loaded liposomes without chitosan coating. Such a system could reduce nephrotoxicity caused by free vancomycin, while enhancing the effect in lung infection. Similarly, nisin encapsulated into cationic liposomes exhibited efficient microbicidal activity against S. mutans [165].
Polymyxin B has been encapsulated into liposomes to target Gram (−) bacterial infections in the sputum of cystic fibrosis patients. Immunocytochemistry revealed a high cell uptake of polymyxin-loaded liposomes, leading to high antimicrobial efficiency toward P. aeruginosa. The bioactivity of liposomal polymyxin B formulation was 4-fold higher compared to AMP alone [166]. Similarly, He et al. encapsulated polymyxin B in a liposomal formulation prepared via the reversed-phase evaporation method. IV injection of liposomal polymyxin exhibited an enhanced pharmacokinetic profile of polymyxin in a pneumonia mice model. Moreover, the efficacy of IV liposomal polymyxin B was significantly higher against P. aeruginosa compared to naked liposomes or polymyxin in solution [167]. The limitations of these studies are the absence of toxicity and hemolysis data, which are critical when polymyxin B is involved.
Still, the success of liposomes depends on understanding the interactions between liposomes and the target site. In this view, Li et al. investigated daptomycin-loaded liposomes for topical skin infections. Both pharmacokinetics and pharmacodynamics were enhanced when using optimal lipid ratios of lecithin to sodium cholate 17:1 (w/w) and a lipid-to-drug ratio of 14:1 (w/w). The liposomal formulation could permeate the skin efficiently and successfully deliver AMP to the site of infection inhibiting S. aureus in mice [168].
Overall, the liposome vectorization offers interesting features for the modulation of pharmacokinetics, which could warrant further investigations for AMP delivery.
4.3. Inorganic NPs
4.3.1. Metallic NPs
The two most used systems of metal NPs are gold (AuNPs) and silver (AgNPs) due to their broad-spectrum activities [169]. These systems offer suitable strategies for delivering AMPs, as both counterparts are usually of low molecular weight. In addition, due to their high surface-to-volume ratio, AMPs can be easily adsorbed to the surface of NPs, resulting in a relatively high AMP loading and efficient delivery to eradicate bacteria. Within this context, esculentin-1a after covalent coupling to AuNPs via a PEG linker showed a 15-fold increased antibacterial activity against P. aeruginosa while being non-toxic to keratinocytes in vitro. Au/esculentin-1 NPs were more resistant to proteolytic degradation with higher potency against P. aeruginosa than the parent peptide and displayed wound-healing properties on keratinocytes in vitro [170]. AMP-coated AuNPs were the first NP system to be proposed for topical application providing antibacterial and wound-healing properties. In another study, AuNPs functionalized with indolicidin were obtained through thiol chemistry by covalently attaching AMP and to AuNPs. The Au/indolicidin nanosystem presented higher efficacy in preventing C. albicans adhesion and eradicated biofilm formation in vitro, as compared to indolicidin alone [171]. The enhanced activity of indolicidin was attributed to the protection from protease degradation conferred by the incorporation into the NPs. In an in vivo study, two AMPs (cecropin and melittin) were conjugated to AuNPs and tested in a sepsis P. aeruginosa- and S. aureus-infected mouse model. AMP–AuNPs (200 µL; 5 mg/mL) lead to decreased bacteremia and significantly lower systemic inflammation than the free peptides. The nanosystem showed higher efficiency than the free peptides in vitro against S. aureus, E.coli, and P. aeruginosa, while being non-hemolytic, non-cytotoxic, and non-immunogenic [172].
A D2A21 AMP encapsulated into Ag nanocomposite exhibited significantly increased antibacterial potency compared to native AMP and AgNPs (MIC: 1 to 3 μg/mL versus 4–6 μg/mL) against P. aeruginosa, S. aureus, and E. coli via membrane damage [40]. An Andersonin-Y1 peptide has been conjugated to AgNPs through a peptide containing a cysteine linker either at the N- or at the C-terminal. Both systems resulted in high activity against K. pneumonia, P. aeruginosa, and Salmonella typhi (Enterbacter spp.), which was nearly 10-fold higher than the parent peptide [140]. The activity of AgNPs against MDR bacteria was enhanced when assisted by the AMP, which disrupted the bacterial membrane, allowing AgNPs to kill bacteria. In another study, the odorranain-A-OA1 (OA1) peptide was coupled to AgNPs through a cysteine in order to stabilize to the nanosystem. AgNP–OA1 conjugates exhibited enhanced bacteria leakage in E. coli and reduced cytotoxicity [173]. Lambadi et al. investigated AgNPs conjugated to polymyxin B and found polymyxin B-AgNPs to display at least 3-fold higher bacterial killing than AgNPs alone. Likewise, AMP–AgNPs showed high potential in destroying biofilm in MDR Vibrio fluvialis and P. aeruginosa [174]. Similarly, Zheng et al. studied Ag nanoclusters conjugated to daptomycin for their antibacterial activity against S. aureus. The nanocluster–AMP system showed higher antibacterial efficiency than the simple mix of daptomycin and Ag nanoclusters. It was suggested that higher antibacterial activity was due to the localization of Ag nanoclusters into the core of the hybrid structure, which could generate reactive oxygen species (ROS) and lead to cell death [175].
Altogether, AMP–metal nanocomposites resulted in synergistic antibacterial effects with enhanced antibacterial activity at low cytotoxicity. Moreover, they proved to be efficient in eradicating biofilms, as they can disrupt the bacterial membrane and finally lead to lysis. However, these nanosystems contain Au or Ag, which are known to be poorly biocompatible and non-biodegradable over long periods of time, and their elimination from the human body is not completely understood [3][176]. AgNPs compounds show very promising results in vitro, although their application can be hindered due to their toxicity seen in vivo [69][72]. As for Au, very recently, Balfourier et al. investigated AuNPs and concluded that Au can be metabolized by mammalian cells. They studied the biotransformation of AuNPs (4 to 22 nm) in primary human fibroblasts for 6 months. First, they could observe that AuNPs follow intracellular degradation induced by ROS that oxidize AuNPs in a size-dependent manner. Second, they demonstrated that gold undergoes a biomineralization process and recrystallizes as nanoleaves (2.5 nm) or so-called aurosomes [177]. Therefore, this brings new insights into the lifecycle of AuNPs and their elimination from the organism.
4.3.2. Carbon Nanotubes, Graphene, Fullerenes
The latest studies in carbon-based nanomaterials, such as graphene, fullerene, carbon quantum dots (CQDs), and carbon nanotubes (CNT) have attracted increasing interest due to their potency in combating infections, while some reports suggest an acceptable toxicity toward mammalian cells and tissues[178][179]. In addition to their antimicrobial potency, carbon-based nanomaterials are very attractive for drug delivery [43]. For instance, CQDs coupled through pyrolysis to L-lysine (Lys-CQDs) and L-arginine (Arg-CQDs) showed effective and selective antibacterial potency with no resistance against E. coli and S. aureus. The in vitro efficacy of the system was due to the induction of intracellular ROS by CQDs. However, no cytotoxicity was reported in mammalian cells. On the contrary, Lys-CQDs and Arg-CQDs promoted the growth of mammalian cells and no hemolysis to red blood cells in vitro. In vivo, both systems significantly inhibited bacterial growth and accelerated wound healing in infected wounds in mice [178]. These systems could lead to different clinical applications of CQDs to eradicate bacteria and promote tissue repair.
Carbon nanotubes (CNTs) are formed by rolled-up tubular shells of graphene. Chaudhary et al. studied the bioconjugation of silver-coated CNTs to AMP TP359 for their antimicrobial potency against MDR Gram (+) (S. aureus and Streptococcus pyogenes) and Gram (−) (Salmonella enterica serovar Typhimurium and Escherichia coli). The bioconjugated system exhibited synergistic antibacterial effects of TP359 and Ag-CNTs and minimal toxicity toward murine macrophages and Hep2 cells within their MIC range [180]. Indolicidin conjugated to CNT and AuNPs exhibited an enhanced potency of indolicidin in an in vitro study. CNT–indolicidin conjugates were found to preserve the activity of indolicidin at a 1000-fold lower concentration. The CNT-carrier activated more pro-inflammatory genes, while AuNP conjugates stimulated the expression of the anti-inflammatory IL-10 gene [181].
Nisin was conjugated to a 3D graphene membrane for the removal of MRSA from water. The graphene system was shown to trap and kill all the bacteria (close to 100%) using a nisin-conjugated membrane. Moreover, the nisin–graphene membrane showed synergistic effects compared to graphene oxide alone or to nisin alone. This time, the multimodal mechanism of action could be observed. Bacteria were captured into the graphene’s foam, which caused stress to the bacterial membrane, allowing nisin to enter, bind easier to bacteria, and kill them efficiently [182]. In addition, Nellore et al. coupled a PGLa (GMASKAGAIAGKIAKVALKAL-NH2) AMP and CNT with a hybrid membrane of glutathione-conjugated carbon nanotube (CNT) bridged three-dimensional (3D) porous graphene oxide for the efficient removal of E. coli. It was found that the system was not only able to capture the bacteria from water, but also to kill E.coli efficiently [183].
A few studies proved that AMPs could be integrated into fullerene C60 nanocrystals using C60 carboxylic groups, which enables covalent coupling between fullerenes and AMPs. Maximin H5 peptide mutants (extracted from Asian toads) were conjugated to fullerenes and showed higher antiviral activities than the maximin H5 peptide [184]. Baron demonstrated that fullerene–peptide conjugates could facilitate transdermal transport in an ex vivo porcine model, whereas the parent peptide did not show any permeation. The fullerene–peptide system could dramatically modify cellular uptake and showed less toxicity to keratinocytes at concentrations below 0.04 mg/mL. They exhibited high efficacy against S. aureus but were inactive against E. coli and C. albicans [185]. Furthermore, AMP–fullerene systems were developed with high bacteriostatic activity against S. aureus and E. coli, exhibiting low MICs of 8 and 64 μM, respectively [186].
Altogether, carbon-based nanosystems show promising results for AMP delivery. Still, concerns of toxicity, induction of granuloma, and potential carcinogenicity in specific models warrant extensive safety studies encompassing a thorough evaluation of their long-term toxicities and pharmacokinetics properties [185][187].
4.4. Smart Nanomaterials
Smart nanomaterials are inspired by natural mechanisms such as those observed in cephalopods (i.e., squid, cuttlefish, or octopus) that respond to external stimuli by changing their color. These so-called “biomimetic” materials can modify their properties in response to external chemical stimuli (e.g., pH, ionic strength) via different interactions. They may offer opportunities to deliver drugs to a specific site or target with an enhanced biological activity.
Hydrogel-forming polysaccharides are widely used in pharmaceutics and cosmetics. They have been used as capping agents for the formulation of AgNPs. Glucuronoxylan (GX) has been used due to its pH-responsive swelling properties upon shifting of the pH from acidic to neutral [188]. This was intended to follow the natural wound pH variation from acidic to slightly alkaline during the different phases of wound healing, due to the imbalance between tissue remodeling and degradation processes [189]. Another antibiotic, fluconazole, has been successfully incorporated into GX hydrogels for drug delivery applications [188]. In another study, AgNPs were prepared using GX polysaccharide to deliver Ag to the infection site. Both GX AgNPs (50 mmol, 5 h sunlight exposure) and Band aid® patches showed comparable wound-healing abilities [190]. The high moisturizing effect of hydrogels at neutral pH is highly beneficial for patients, preventing bandage adhesion. Moreover, incorporated AgNPs showed high bactericidal and fungicidal activity against S. epidermidis, E. coli, P. aeruginosa, B. subtilis, S. aureus and A. niger, P. notatum, R. stolonifer, and A. odontolyticus. As well, rapid wound-healing tendency was seen when using AgNPs–GX hydrogels in rabbits (Figure 8).
Figure 8. Preparation of smart wound dressing based on AgNPs–GX (gold nanoparticles–glucuronoxylan) hydrogels and their application for wound treatment. Modified from [190].
Electrospun hybrid polyacrilonitrile (PAN) nanofiber mats have been developed for wound healing, possessing the ability to monitor in real-time the H2O2 level in the wound [191]. High H2O2 concentration in wounds leads to delayed healing, hindering connective tissue formation [192]. The PAN smart dressings could sense H2O2 concentration through fluorescence changes upon UV irradiation, indicating the level of bacterial infection and the time to change the mats. Such smart dressing approaches could help monitor in vivo wounds in an inflammatory model. Real-time information could help avoid infection and shorten the healing time.
Nanofibrous electrospun membranes were prepared by loading an antibiotic, such as nitrofurazone, into Eudragit® S100 (ES 100) polymer solution as potential smart nanomaterial for skin infection treatment. ES100 is a pH-responsive polymer that dissolves at pH higher than 7, thus triggering the release of the antibacterial agent [189]. Rezaei et al. have prepared thermo-responsive chitosan (TCTS) hydrogels and loaded them with piscidin-1 AMP at 16 μg/mL to obtain efficient antibacterial wound dressing against resistant strains of A. baumannii. The use of TCTS hydrogels as a carrier for piscidin-1 showed to be advantageous with high antibacterial activity and an absence of toxicity to mammal cells [193].
Panzaras et al. [194] developed “bandages with a voice” using a pyranine–benzalkonium ion pair loaded into wound dressings that are able to detect pH changes in the range of 5.5–7.5, based on the UV-induced fluorescence color change of a dye. The pH of the healing wound is around pH 5–6, while the pH in chronic wounds fluctuates between 7 and 8, indicating bacterial infection (Figure 9). These kinds of dressings are practical and very much needed for monitoring the status of the wounds, although the pH sensitivity range is quite narrow, and a UV lamp is required.
Figure 9. Understanding wound status: elevated or extremely low pH in wound bed can be a sign of infection. Modified from [194].
A very sound strategy to design smart nanomaterials has been proposed by Lombardi et al. [195]. Their system could incorporate conventional antibiotics while being stimuli responsive, so at a certain pH, it would release the antibiotic. An AMP developed from innate immunity peptides found in the hagfish, WMR, was linked to an aliphatic residue having a lipidic tail with pH-dependent properties. For instance, at pH 10, the peptides self-assemble into stable nanofibers that are highly active against C. albicans, P. aeruginosa, and biofilm bacteria, while at pH 3 and 7, they do not self-assemble. These nanostructures hold great potential as stimuli-responsive materials for infection treatment in the wounds.
RADA16, a self-assembling peptide that is able to form 3D-hydrogel in response to pH changes, has received considerable attention due to its remarkable properties to deliver different proteins and drugs, to promote tissue regeneration, and to influence cell behavior [196]. It is worth noting that proteins can preserve their original structure and function inside RADA16 hydrogels. RADA16 was functionalized with a RGDS fibronectin moiety (RADA16-RGD) or with a FPGERGVEGPGP collagen-1 (RADA16-FGP) motif via two glycine residues to ensure flexibility to the functional groups. Functionalized RADA16-FGP was shown to promote the migration of keratinocytes and fibroblasts significantly, while RADA16-RGD had no effect on cells [197]. In addition, RADA16-RGD added with TGF-β1 was used for growing bone mesenchymal cells from rats over the course of 2 weeks. The results showed that RADA16 peptide hydrogels were able to release active TGF-β1 growth factor and significantly influence the proliferation of stem cells [198].
These types of smart biomaterials could induce diverse cell responses within a regenerative scaffold. Therefore, the incorporation of AMP in such smart and wound environment-responsive systems could be successfully used to promote not only wound healing but to protect wounds against microbial infections.
Overall, incorporating AMP into different delivery nanosystems showed an improved bioactivity with higher rate of bacterial killing while reducing their toxicity. Very surprisingly, Håkansson et al. loaded DPK-060 into three different nanosystems: lipid nanocapsules (LNCs), nanocubes, and polaxamer gels compared to the parent DPK-060 peptide in acetate buffer. No enhanced effect was found when encapsulating DPK-060 in any of the formulations, except when including AMP in polaxamer gels [199]. This points out the need for a specific optimization of the carrier for a given peptide through activity and toxicity testing.
5. Discussion and Further Insights
AMPs have raised attention due to their potent activity along with many advantages, such as broad activity not only against common strains, but MDR bacteria as well, which is of utmost interest nowadays. They show relatively low bacterial resistance and high activity against biofilm-forming bacteria, especially critical in implant- or device-related infections, although very little is yet known about resistance mechanisms developed by biofilms.
The main limitations for the use of AMPs are summarized in Table 3. Poor stability may be overcome by several approaches, as mentioned in Section 3. Cytotoxicity and hemolysis are critical issues that need to be addressed, for instance by conjugation, PEGylation, or formulation as detailed above. Finally, production costs might be an additional barrier to the clinical use of AMPs.
| Advantages | Disadvantages |
|---|---|
| Several AMPs show broad and simultaneous activities against bacteria, fungi, viruses, and in case of infection with multiple microorganisms, one AMP could be efficient to overcome this issue. | AMPs are susceptible to proteolytic degradation, which leads to the loss of biological activity. |
| Natural AMPs are already found in high doses at the site of infection. | Some AMPs can be toxic to mammal cells at high concentrations. |
| Some AMPs have wound-healing and angiogenesis promotion properties, which are essential in case of hard-to-heal and infected wounds, such as diabetes and foot ulcer. | AMPs can induce hypersensitivity reactions after application. |
| AMPs show as well anti-inflammatory properties by modulating immune cytokines, which are responsible for the inflammatory response. | AMPs can be influenced by pH variation and at low concentration of salt can be destabilized, leading to loss of activity. |
| AMPs exhibit therapeutic antimicrobial activity at extremely low concentrations in the microscale and sometimes nanoscale range. | AMP’s production and purification costs are high. |
| Resistance to AMPs is very low and only in a limited number of AMPs. | |
| AMPs showed to be time-efficient; some AMPs can act within a few seconds to a few minutes. | |
| AMPs inhibit biofilm formation, which is especially important to prevent bacterial growth on medical devices. | |
| AMPs show synergism when chemically coupled to polymers, encapsulated into different delivery systems or simultaneously applied with antimicrobial agents. | |
| AMPs can act synergistically with antibiotics. |
Clinical research on AMPs may bring insight into future developments. Only a minority of AMPs has obtained FDA approval. Neosporin® (gramicidin), Cubicin® (daptomycin), Vancocin® HCl (vancomycin), Orbactiv® (oritavancin), and Dalvance™ (dalbavancin) have been approved for the treatment of bacterial skin infections or wounds [44]. Most of the currently approved AMPs, except colistin, are used to treat Gram (+) bacteria, and a few of them have low oral bioavailability or poor intestinal mucosa penetration and fast enzyme degradation [200]. On the other hand, oral Vancomycin® HCl administration in patients with severe colitis and renal insufficiency leads to severe intestine mucosal damage due to extremely high bioavailability [201]. Therefore, drug monitoring is highly suggested in cases of high risk, such as colitis or renal insufficiency. Moreover, AMPs that are efficient to treat Gram (−) bacteria are now emerging.
Although vancomycin has been FDA approved and used to treat MRSA- and C. difficile-colitis, it has been found to induce peripheral rash, anaphylaxis, “red man syndrome”, ototoxicity, and nephrotoxicity [201][202]. Daptomycin is used as an alternative to vancomycin and has been FDA approved for MRSA bloodstream infections. On the other side, colistin and daptomycin are considered as extremely important AMPs for the treatment of severe human diseases and other diseases transmissible from non-human sources to humans [203]. However, colistin may cause severe neuro- and nephrotoxicity and is used only as a “last-resort” antibiotic [203]. Telavancin, another derivative of vancomycin, has been approved for MDR infections, especially for osteomyelitis treatment, but it leads to adverse effects, such as nausea, vomiting, diarrhea, and metallic taste [204]. Oritavancin has fewer side effects than vancomycin and was found to be safe in patients with osteomyelitis even when administering multiple doses a day [205]. These clinical outcomes suggest that it is essential to check the selectivity when designing a new AMP with increased bioactivity and reduced toxicity. Progress in AMP design, testing, and vectorization suggests that the number of AMPs will be increasing in the near future to combat MDR bacteria in wound infections and biofilm-related infections.
Toxicity to mammalian cells and AMP short half-life are main factors limiting their clinical application. Therefore, the topical delivery of AMPs, especially to the wounds, is the most common route of administration. Still, AMPs could be easily degraded by wound exudates. Therefore, the different delivery strategies described herein, such as conjugation of the AMPs to polymers, encapsulation into different nanosystems, and smart nanomaterials or new technologies such as CRISPR-Cas9 could be used to overcome the hurdles of AMPs alone while maintaining high antimicrobial activity. So far, there is no commercial formulation available specifically for the topical application of AMPs.
Advanced gene engineering tools for AMP synthesis, such as Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR)-associated protein (CRISPR-Cas, a RNA-guided genome editing system) and Zinc-Finger Nucleases (ZNFs), are new approaches recently exploited that are still not fully understood but deserve future research and investigations [206]. Such modern tools can be used to specifically target AMR bacterial strains that carry those resistant genes and re-sensitize bacteria to antibiotic treatment [207]. For example, the group of Lemaitre has been using the CRISPR-Cas9 endonuclease to delete 14 AMP genes of Drosophila. The results showed that the obtained AMP mutants were efficient against Gram-negative bacteria (e.g., E. coli and Enterobacter species) and fungi in Drosophila [208]. The same group could observe that only Diptericins could resist the infection of Providencia rettgeri [208], which may correlate with the polymorphism in Drosophila [209]. CRISPR-Cas9 mediated AMP gene knockout could serve as an example of how to engineer novel and efficient AMPs for patients lacking a specific AMP to prevent certain infections.
Choudhary et al. 2019 [210] and Wei et al. 2019 [211] have used the endogenous CRISPR-Cas system as an efficient tool against Mycobacterium tuberculosis, which has developed multidrug-resistance, even against the most potent antibiotic rifampicin. In other studies, CRISPR-Cas9 was successfully delivered to selectively kill MDR bacteria E. coli [212], and S. aureus [213] using a phagemid (plasmid packed in a phage capsid)-mediated system by removing plasmids carrying AMR genes and, in turn, effectively re-sensitizing bacteria to antibiotics. Moreover, both the phagemid-mediated delivery of CRISPR-Cas9 was shown to be highly effective in vivo in E. coli-infected Galleria mellonella larvae [212] and S. aureus in a mouse skin colonization model [213].
Although CRISPR-Cas proved to be an efficient tool to enhance AMP antimicrobial activity as antimicrobials, it is very important that the protospacer adjacent motif (PAM) sequences and Cas proteins are fully characterized [214]. In addition, the use of recombinant microbial platforms for genetically tailoring AMPs leads very often to low yield and poor quality of the final product [206]. Therefore, a very limited number of genetically modified AMPs [215] have reached clinical trials; thus, these AMPs are less marketed, which has been described in detail by Kosikowska and Lesner.
With respect to delivery strategies, general trends may be distinguished despite the lack of comparative studies in the field. A salient feature is the ability of several delivery strategies to protect the peptide from degradation and/or to sustain its activity at the diseased site. This is true for several polymer conjugates, but also for polymer nanogels. Chitosan stands out as a promising biopolymer in this respect. Nanotechnological platforms may also be used to improve solubility and bioavailability. As a final topical formulation, these may be integrated into a polymer scaffold to provide both controlled peptide delivery and additionally promote wound healing.
Still, the safety of these systems needs careful assessment, which should include their hemolytic activity and potential side effects. In this respect, more recent approaches relying on metallic nanoparticles, nanotubes, or fullerenes warrant further investigation to confirm a beneficial efficacy/toxicity balance.
Finally, combined strategies, such as the use of antibiotics for synergistic action or the combination with antibiotic resistance breakers might be required to further potentialize anti-infective therapy.
Altogether, it is important to design new AMPs with high antibacterial activity, but it is essential to optimize them via proposed strategies to efficiently and safely deliver them at the site of infection. AMPs remain one of the most promising classes of antimicrobial agents that brings hope to overcome AMR and biofilm-related infections.
References
- Patrulea, V.; Ostafe, V.; Borchard, G.; Jordan, O. Chitosan as a starting material for wound healing applications. European Journal of Pharmaceutics and Biopharmaceutics 2015, 97, 417-426. https://doi.org/10.1016/j.ejpb.2015.08.004.
- Singh, M.; Jonnalagadda, S. Advances in bioprinting using additive manufacturing. European Journal of Pharmaceutical Sciences 2020, 143, 105167. https://doi.org/10.1016/j.ejps.2019.105167.
- Thapa, R.K.; Diep, D.B.; Tønnesen, H.H. Topical antimicrobial peptide formulations for wound healing: Current developments and future prospects. Acta Biomaterialia 2020, 103, 52-67. https://doi.org/10.1016/j.actbio.2019.12.025.
- Gieringer, M.; Gosepath, J.; Naim, R. Radiotherapy and wound healing: principles, management and prospects (review). Oncology reports 2011, 26(2), 299-307. 10.3892/or.2011.1319.
- Abdel-Sayed, P.; Kaeppeli, A.; Siriwardena, T.; Darbre, T.; Perron, K.; Jafari, P.; Reymond, J.-L.; Pioletti, D.P.; Applegate, L.A. Anti-Microbial Dendrimers against Multidrug-Resistant P. aeruginosa Enhance the Angiogenic Effect of Biological Burn-wound Bandages. Sci Rep 2016, 6(1), 22020. https://doi.org/10.1038/srep22020.
- Patrulea, V.; Younes, I.; Jordan, O.; Borchard, G. Chitosan-based systems for controlled delivery of antimicrobial peptides for biomedical application. In: Functional chitosan: drug delivery and biomedical applications, ed.; Jana, S.; Jana, S., editors; Springer, Singapore. 2019; 415-455.
- Omar, A.; Wright, J.B.; Schultz, G.; Burrell, R.; Nadworny, P. Microbial Biofilms and Chronic Wounds. Microorganisms 2017, 5(1). https://doi.org/10.3390/microorganisms5010009.
- Lombardi, L.; Falanga, A.; Del Genio, V.; Galdiero, S. A New Hope: Self-Assembling Peptides with Antimicrobial Activity. Pharmaceutics 2019, 11(4), 166. https://doi.org/10.3390/pharmaceutics11040166.
- Seppanen, E.J.; Thornton, R.B.; Corscadden, K.J.; Granland, C.M.; Hibbert, J.; Fuery, A.; Wiertsema, S.P.; Vijayasekaran, S.; Coates, H.L.; Jacoby, P.; Currie, A.; Richmond, P.C.; Kirkham, L.S. High concentrations of middle ear antimicrobial peptides and proteins and proinflammatory cytokines are associated with detection of middle ear pathogens in children with recurrent acute otitis media. PloS one 2019, 14(12), e0227080. https://doi.org/10.1371/journal.pone.0227080.
- Rios, A.C.; Moutinho, C.G.; Pinto, F.C.; Del Fiol, F.S.; Jozala, A.; Chaud, M.V.; Vila, M.M.D.C.; Teixeira, J.A.; Balcão, V.M. Alternatives to overcoming bacterial resistances: State-of-the-art. Microbiological Research 2016, 191, 51-80. https://doi.org/10.1016/j.micres.2016.04.008.
- Hincapié, O.; Giraldo, P.; Orduz, S. In silico design of polycationic antimicrobial peptides active against Pseudomonas aeruginosa and Staphylococcus aureus. Antonie van Leeuwenhoek 2018, 111(10), 1871-1882. 10.1007/s10482-018-1080-2.
- Gull, S.; Shamim, N.; Minhas, F. AMAP: Hierarchical multi-label prediction of biologically active and antimicrobial peptides. Computers in Biology and Medicine 2019, 107, 172-181. https://doi.org/10.1016/j.compbiomed.2019.02.018.
- Haney, E.F.; Hancock, R.E.W. Peptide design for antimicrobial and immunomodulatory applications. Biopolymers 2013, 100(6), 572-583. 10.1002/bip.22250.
- Meher, P.K.; Sahu, T.K.; Saini, V.; Rao, A.R. Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou's general PseAAC. Sci Rep 2017, 7, 42362-42362. 10.1038/srep42362.
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L.; Cherkasov, A.; Seleem, M.N.; Pinilla, C.; De La Fuente-Nunez, C.; Lazaridis, T.; Dai, T.; Houghten, R.A.; Hancock, R.E.W.; Tegos, G.P. The value of antimicrobial peptides in the age of resistance. The Lancet Infectious Diseases 2020. https://doi.org/10.1016/S1473-3099(20)30327-3.
- Porto, W.F.; Pires, A.S.; Franco, O.L. Computational tools for exploring sequence databases as a resource for antimicrobial peptides. Biotechnology Advances 2017, 35(3), 337-349. https://doi.org/10.1016/j.biotechadv.2017.02.001.
- Torres, M.D.T.; Sothiselvam, S.; Lu, T.K.; De La Fuente-Nunez, C. Peptide Design Principles for Antimicrobial Applications. Journal of Molecular Biology 2019, 431(18), 3547-3567. https://doi.org/10.1016/j.jmb.2018.12.015.
- Fleming, A.; Wright, A.E. On a remarkable bacteriolytic element found in tissues and secretions. Proc. R. Soc. B 1922, 93(653), 306-317. https://doi.org/10.1098/rspb.1922.0023.
- Fleming, A. On the Antibacterial Action of Cultures of a Penicillium, with Special Reference to their Use in the Isolation of B. influenzæ. Br. J. Exp. Pathol. 1929, 10(3), 226-236.
- APD (The Antimicrobial Peptide Database) http://aps.unmc.edu/AP/main.php (accessed on 05/03/2020).
- DRAMP (Data repository of antimicrobial peptides) http://dramp.cpu-bioinfor.org/browse/ (accessed 05/03/2020).
- Kang, X.; Dong, F.; Shi, C.; Liu, S.; Sun, J.; Chen, J.; Li, H.; Xu, H.; Lao, X.; Zheng, H. DRAMP 2.0, an updated data repository of antimicrobial peptides. Scientific Data 2019, 6(1), 148. https://doi.org/10.1038/s41597-019-0154-y.
- Koo, H.B.; Seo, J. Antimicrobial peptides under clinical investigation. Peptide Sci. 2019, 111(5), e24122. https://doi.org/10.1002/pep2.24122.
- Nuti, R.; Goud, N.S.; Saraswati, A.P.; Alvala, R.; Alvala, M. Antimicrobial Peptides: A Promising Therapeutic Strategy in Tackling Antimicrobial Resistance. Curr. Med. Chem. 2017, 24(38), 4303-4314. https://doi.org/10.2174/0929867324666170815102441.
- Boto, A.; Perez De La Lastra, J.M.; Gonzalez, C.C. The Road from Host-Defense Peptides to a New Generation of Antimicrobial Drugs. Molecules 2018, 23(2). https://doi.org/10.3390/molecules23020311.
- Ho, Y.-H.; Shah, P.; Chen, Y.-W.; Chen, C.-S. Systematic Analysis of Intracellular-targeting Antimicrobial Peptides, Bactenecin 7, Hybrid of Pleurocidin and Dermaseptin, Proline-Arginine-rich Peptide, and Lactoferricin B, by Using Escherichia coli Proteome Microarrays. Mol. Cell. Proteomics 2016, 15(6), 1837-1847. https://doi.org/10.1074/mcp.M115.054999.
- Rowe-Magnus, D.A.; Kao, A.Y.; Prieto, A.C.; Pu, M.; Kao, C. Cathelicidin Peptides Restrict Bacterial Growth via Membrane Perturbation and Induction of Reactive Oxygen Species. mBio 2019, 10(5), e02021-02019. https://doi.org/10.1128/mBio.02021-19.
- Cho, J.; Hwang, I.-S.; Choi, H.J.; Ji Hong, H.; Hwang, J.-S.; Lee, D.G. The novel biological action of antimicrobial peptides via apoptosis induction. J. Microbiol. Biotechnol. 2012, 22(11), 1457-1466. http://dx.doi.org/10.4014/jmb.1205.05041.
- Mangoni, M.L.; Mcdermott, A.M.; Zasloff, M. Antimicrobial peptides and wound healing: biological and therapeutic considerations. Experimental dermatology. 2016, 25(3), 167-173. https://doi.org/10.1111/exd.12929.
- Steinstraesser, L.; Hirsch, T.; Schulte, M.; Kueckelhaus, M.; Jacobsen, F.; Mersch, E.A.; Stricker, I.; Afacan, N.; Jenssen, H.; Hancock, R.E.; Kindrachuk, J. Innate defense regulator peptide 1018 in wound healing and wound infection. PloS one 2012, 7(8), e39373. https://doi.org/10.1371/journal.pone.0039373.
- Hirsch, T.; Spielmann, M.; Zuhaili, B.; Fossum, M.; Metzig, M.; Koehler, T.; Steinau, H.U.; Yao, F.; Onderdonk, A.B.; Steinstraesser, L.; Eriksson, E. Human beta-defensin-3 promotes wound healing in infected diabetic wounds. The journal of gene medicine 2009, 11(3), 220-228. https://doi.org/10.1002/jgm.1287.
- Kolar, S.S.; Mcdermott, A.M. Role of host-defence peptides in eye diseases. Cellular and Molecular Life Sciences 2011, 68(13), 2201. https://doi.org/10.1007/s00018-011-0713-7.
- Shaykhiev, R.; Beisswenger, C.; Kandler, K.; Senske, J.; Puchner, A.; Damm, T.; Behr, J.; Bals, R. Human endogenous antibiotic LL-37 stimulates airway epithelial cell proliferation and wound closure. American journal of physiology. Lung cellular and molecular physiology 2005, 289(5), L842-848. https://doi.org/10.1152/ajplung.00286.2004.
- Gomes, A.; Teixeira, C.; Ferraz, R.; Prudencio, C.; Gomes, P. Wound-Healing Peptides for Treatment of Chronic Diabetic Foot Ulcers and Other Infected Skin Injuries. Molecules 2017, 22(10). https://doi.org/10.3390/molecules22101743.
- Teirlinck, E.; Samal, S.K.; Coenye, T.; Braeckmans, K. Chapter 3 - Penetrating the Bacterial Biofilm: Challenges for Antimicrobial Treatment. In: Functionalized Nanomaterials for the Management of Microbial Infection, ed.; Boukherroub, R.; Szunerits, S.; Drider, D., editors; Elsevier.Boston, 2017; 49-76.
- Nishikawa, T.; Nakagami, H.; Maeda, A.; Morishita, R.; Miyazaki, N.; Ogawa, T.; Tabata, Y.; Kikuchi, Y.; Hayashi, H.; Tatsu, Y.; Yumoto, N.; Tamai, K.; Tomono, K.; Kaneda, Y. Development of a novel antimicrobial peptide, AG-30, with angiogenic properties. Journal of Cellular and Molecular Medicine 2009, 13(3), 535-546. https://doi.org/10.1111/j.1582-4934.2008.00341.x.
- Nakagami, H.; Nishikawa, T.; Tamura, N.; Maeda, A.; Hibino, H.; Mochizuki, M.; Shimosato, T.; Moriya, T.; Morishita, R.; Tamai, K.; Tomono, K.; Kaneda, Y. Modification of a novel angiogenic peptide, AG30, for the development of novel therapeutic agents. Journal of Cellular and Molecular Medicine 2012, 16(7), 1629-1639. https://doi.org/10.1111/j.1582-4934.2011.01406.x.
- Liu, H.; Mu, L.; Tang, J.; Shen, C.; Gao, C.; Rong, M.; Zhang, Z.; Liu, J.; Wu, X.; Yu, H.; Lai, R. A potential wound healing-promoting peptide from frog skin. The International Journal of Biochemistry and Cell Biology 2014, 49, 32-41. https://doi.org/10.1016/j.biocel.2014.01.010.
- Mu, L.; Tang, J.; Liu, H.; Shen, C.; Rong, M.; Zhang, Z.; Lai, R. A potential wound-healing-promoting peptide from salamander skin. The FASEB Journal 2014, 28(9), 3919-3929. https://doi.org/10.1096/fj.13-248476.
- Tang, J.; Liu, H.; Gao, C.; Mu, L.; Yang, S.; Rong, M.; Zhang, Z.; Liu, J.; Ding, Q.; Lai, R. A small peptide with potential ability to promote wound healing. PloS one 2014, 9(3), e92082. https://doi.org/10.1371/journal.pone.0092082.
- Ma, Z.; Han, J.; Chang, B.; Gao, L.; Lu, Z.; Lu, F.; Zhao, H.; Zhang, C.; Bie, X. Membrane-Active Amphipathic Peptide WRL3 with in Vitro Antibiofilm Capability and in Vivo Efficacy in Treating Methicillin-Resistant Staphylococcus aureus Burn Wound Infections. ACS Infectious Diseases 2017, 3(11), 820-832. https://doi.org/10.1021/acsinfecdis.7b00100.
- Huang, H.-N.; Pan, C.-Y.; Wu, H.-Y.; Chen, J.-Y. Antimicrobial peptide Epinecidin-1 promotes complete skin regeneration of methicillin-resistant Staphylococcus aureus-infected burn wounds in a swine model. Oncotarget 2017, 8(13), 21067-21080. https://doi.org/10.18632/oncotarget.15042.
- Nordström, R.; Malmsten, M. Delivery systems for antimicrobial peptides. Adv. Colloid Interface Sci. 2017, 242, 17-34. https://doi.org/10.1016/j.cis.2017.01.005.
- Chen, C.H.; Lu, T.K. Development and Challenges of Antimicrobial Peptides for Therapeutic Applications. Antibiotics 2020, 9(1). https://doi.org/10.3390/antibiotics9010024.
- Song, D.W.; Kim, S.H.; Kim, H.H.; Lee, K.H.; Ki, C.S.; Park, Y.H. Multi-biofunction of antimicrobial peptide-immobilized silk fibroin nanofiber membrane: Implications for wound healing. Acta Biomaterialia 2016, 39, 146-155. https://doi.org/10.1016/j.actbio.2016.05.008.
- Gunasekera, S.; Muhammad, T.; Strömstedt, A.A.; Rosengren, K.J.; Göransson, U. Backbone Cyclization and Dimerization of LL-37-Derived Peptides Enhance Antimicrobial Activity and Proteolytic Stability. Front Microbiol 2020, 11. https://doi.org/10.3389/fmicb.2020.00168.
- Paranjape, S.M.; Lauer, T.W.; Montelaro, R.C.; Mietzner, T.A.; Vij, N. Modulation of proinflammatory activity by the engineered cationic antimicrobial peptide WLBU-2. F1000Res 2013, 2, 36-36. 10.12688/f1000research.2-36.v1.
- Lin, Q.; Deslouches, B.; Montelaro, R.C.; Di, Y.P. Prevention of ESKAPE pathogen biofilm formation by antimicrobial peptides WLBU2 and LL37. International Journal of Antimicrobial Agents 2018, 52(5), 667-672. https://doi.org/10.1016/j.ijantimicag.2018.04.019.
- Orlov, D.S.; Shamova, O.V.; Eliseev, I.E.; Zharkova, M.S.; Chakchir, O.B.; Antcheva, N.; Zachariev, S.; Panteleev, P.V.; Kokryakov, V.N.; Ovchinnikova, T.V.; Tossi, A. Redesigning Arenicin-1, an Antimicrobial Peptide from the Marine Polychaeta Arenicola marina, by Strand Rearrangement or Branching, Substitution of Specific Residues, and Backbone Linearization or Cyclization. Mar Drugs 2019, 17(6), 376. https://doi.org/10.3390/md17060376.
- Elliott, A.G.; Huang, J.X.; Neve, S.; Zuegg, J.; Edwards, I.A.; Cain, A.K.; Boinett, C.J.; Barquist, L.; Lundberg, C.V.; Steen, J.; Butler, M.S.; Mobli, M.; Porter, K.M.; Blaskovich, M.a.T.; Lociuro, S.; Strandh, M.; Cooper, M.A. An amphipathic peptide with antibiotic activity against multidrug-resistant Gram-negative bacteria. Nature communications 2020, 11(1), 3184-3184. 10.1038/s41467-020-16950-x.
- Kunda, N.K. Antimicrobial peptides as novel therapeutics for non-small cell lung cancer. Drug Discovery Today 2020, 25(1), 238-247. https://doi.org/10.1016/j.drudis.2019.11.012.
- Hammami, R.; Fernandez, B.; Lacroix, C.; Fliss, I. Anti-infective properties of bacteriocins: an update. Cellular and Molecular Life Sciences 2013, 70(16), 2947-2967. https://doi.org/10.1007/s00018-012-1202-3.
- Dover, S.E.; Aroutcheva, A.A.; Faro, S.; Chikindas, M.L. Safety study of an antimicrobial peptide lactocin 160, produced by the vaginal Lactobacillus rhamnosus. Infect Dis Obstet Gynecol 2007, 2007, 78248-78248. 10.1155/2007/78248.
- Meade, E.; Slattery, M.A.; Garvey, M. Bacteriocins, Potent Antimicrobial Peptides and the Fight against Multi Drug Resistant Species: Resistance Is Futile? Antibiotics 2020, 9(1), 32. https://doi.org/doi:10.3390/antibiotics9010032.
- Sherwood, E.J.; Bibb, M.J. The antibiotic planosporicin coordinates its own production in the actinomycete Planomonospora alba. Proc Natl Acad Sci U S A 2013, 110(27), E2500-E2509. https://doi.org/10.1073/pnas.1305392110.
- Castiglione, F.; Lazzarini, A.; Carrano, L.; Corti, E.; Ciciliato, I.; Gastaldo, L.; Candiani, P.; Losi, D.; Marinelli, F.; Selva, E.; Parenti, F. Determining the Structure and Mode of Action of Microbisporicin, a Potent Lantibiotic Active Against Multiresistant Pathogens. Chemistry & Biology 2008, 15(1), 22-31. https://doi.org/10.1016/j.chembiol.2007.11.009.
- Ghosh, C.; Sarkar, P.; Issa, R.; Haldar, J. Alternatives to Conventional Antibiotics in the Era of Antimicrobial Resistance. Trends in Microbiology 2019, 27(4), 323-338. https://doi.org/10.1016/j.tim.2018.12.010.
- Hara, S.; Mukae, H.; Sakamoto, N.; Ishimoto, H.; Amenomori, M.; Fujita, H.; Ishimatsu, Y.; Yanagihara, K.; Kohno, S. Plectasin has antibacterial activity and no affect on cell viability or IL-8 production. Biochemical and Biophysical Research Communications 2008, 374(4), 709-713. https://doi.org/10.1016/j.bbrc.2008.07.093.
- Mahlapuu, M.; Håkansson, J.; Ringstad, L.; Björn, C. Antimicrobial Peptides: An Emerging Category of Therapeutic Agents. Frontiers in cellular and infection microbiology 2016, 6, 194-194. https://doi.org/10.3389/fcimb.2016.00194.
- Muchintala, D.; Suresh, V.; Raju, D.; Sashidhar, R.B. Synthesis and characterization of cecropin peptide-based silver nanocomposites: Its antibacterial activity and mode of action. Materials Science and Engineering: C 2020, 110, 110712. https://doi.org/10.1016/j.msec.2020.110712.
- Van Groenendael, R.; Kox, M.; Leijte, G.; Koeneman, B.; Gerretsen, J.; Van Eijk, L.; Pickkers, P. A randomized double-blind, placebo-controlled clinical phase IIa trial on safety, immunomodulatory effects and pharmacokinetics of EA-230 during experimental human endotoxaemia. Br J Clin Pharmacol 2019, 85(7), 1559-1571. https://doi.org/10.1111/bcp.13941.
- Hazam, P.K.; Goyal, R.; Ramakrishnan, V. Peptide based antimicrobials: Design strategies and therapeutic potential. Progress in Biophysics and Molecular Biology 2019, 142, 10-22. https://doi.org/10.1016/j.pbiomolbio.2018.08.006.
- Han, J.; Jyoti, M.A.; Song, H.-Y.; Jang, W.S. Antifungal Activity and Action Mechanism of Histatin 5-Halocidin Hybrid Peptides against Candida ssp. PloS one 2016, 11(2), e0150196-e0150196. 10.1371/journal.pone.0150196.
- Costa, F.; Maia, S.; Gomes, J.; Gomes, P.; Martins, M.C.L. Characterization of hLF1–11 immobilization onto chitosan ultrathin films, and its effects on antimicrobial activity. Acta Biomaterialia 2014, 10(8), 3513-3521. https://doi.org/10.1016/j.actbio.2014.02.028.
- Bruni, N.; Capucchio, M.T.; Biasibetti, E.; Pessione, E.; Cirrincione, S.; Giraudo, L.; Corona, A.; Dosio, F. Antimicrobial Activity of Lactoferrin-Related Peptides and Applications in Human and Veterinary Medicine. Molecules 2016, 21(6). 10.3390/molecules21060752.
- Overhage, J.; Campisano, A.; Bains, M.; Torfs, E.C.W.; Rehm, B.H.A.; Hancock, R.E.W. Human Host Defense Peptide LL-37 Prevents Bacterial Biofilm Formation. Infection and Immunity 2008, 76(9), 4176-4182. 10.1128/iai.00318-08.
- US National Library of Medicine. A Phase II Study to Evaluate the Efficacy and Safety of Two Doses of LTX-109 in Impetigo https://clinicaltrials.gov/ct2/show/NCT01803035 (accessed on 26/03/2020).
- Saravolatz, L.D.; Pawlak, J.; Johnson, L.; Bonilla, H.; Saravolatz, L.D., 2nd; Fakih, M.G.; Fugelli, A.; Olsen, W.M. In vitro activities of LTX-109, a synthetic antimicrobial peptide, against methicillin-resistant, vancomycin-intermediate, vancomycin-resistant, daptomycin-nonsusceptible, and linezolid-nonsusceptible Staphylococcus aureus. Antimicrob Agents Chemother 2012, 56(8), 4478-4482. 10.1128/AAC.00194-12.
- Yasir, M.; Dutta, D.; Hossain, K.R.; Chen, R.; Ho, K.K.K.; Kuppusamy, R.; Clarke, R.J.; Kumar, N.; Willcox, M.D.P. Mechanism of Action of Surface Immobilized Antimicrobial Peptides Against Pseudomonas aeruginosa. Front. Microbiol. 2020, 10(3053). https://doi.org/10.3389/fmicb.2019.03053.
- Yasir, M.; Dutta, D.; Willcox, M.D.P. Comparative mode of action of the antimicrobial peptide melimine and its derivative Mel4 against Pseudomonas aeruginosa. Sci Rep 2019, 9(1), 7063. 10.1038/s41598-019-42440-2.
- Czyzewski, A.M.; Jenssen, H.; Fjell, C.D.; Waldbrook, M.; Chongsiriwatana, N.P.; Yuen, E.; Hancock, R.E.; Barron, A.E. In Vivo, In Vitro, and In Silico Characterization of Peptoids as Antimicrobial Agents. PloS one 2016, 11(2), e0135961. https://doi.org/10.1371/journal.pone.0135961.
- Vila, J.; Moreno-Morales, J.; Ballesté-Delpierre, C. Current landscape in the discovery of novel antibacterial agents. Clinical Microbiology and Infection 2019. https://doi.org/10.1016/j.cmi.2019.09.015.
- Du, H.; Puri, S.; Mccall, A.; Norris, H.L.; Russo, T.; Edgerton, M. Human Salivary Protein Histatin 5 Has Potent Bactericidal Activity against ESKAPE Pathogens. Frontiers in Cellular and Infection Microbiology 2017, 7(41). 10.3389/fcimb.2017.00041.
- US National Library of Medicine. Phase III efficacy and safety study of AB103 in the treatment of patients with necrotizing soft tissue infections (ACCUTE) https://clinicaltrials.gov/ct2/show/NCT02469857 (accessed on 26/03/2020)
- De La Cruz, M.; González, I.; Parish, C.A.; Onishi, R.; Tormo, J.R.; Martín, J.; Peláez, F.; Zink, D.; El Aouad, N.; Reyes, F.; Genilloud, O.; Vicente, F. Production of Ramoplanin and Ramoplanin Analogs by Actinomycetes. Front Microbiol 2017, 8(343). https://doi.org/10.3389/fmicb.2017.00343.
- Cheng, M.; Huang, J.X.; Ramu, S.; Butler, M.S.; Cooper, M.A. Ramoplanin at bactericidal concentrations induces bacterial membrane depolarization in Staphylococcus aureus. Antimicrob Agents Chemother 2014, 58(11), 6819-6827. 10.1128/AAC.00061-14.
- Bienvenu, A.-L.; Pradat, P.; Guerin, C.; Aubrun, F.; Fellahi, J.-L.; Friggeri, A.; Guichon, C.; Hernu, R.; Menotti, J.; Monard, C.; Paulus, S.; Rimmele, T.; Piriou, V.; Chidiac, C.; Argaud, L.; Leboucher, G. Evaluation of first-line therapies for the treatment of candidemia in ICU patients: A propensity score analysis. Int. J. Infect. Dis. 2020, 93, 15-21. https://doi.org/10.1016/j.ijid.2020.01.037.
- Pfaller, M.A.; Diekema, D.J.; Ostrosky-Zeichner, L.; Rex, J.H.; Alexander, B.D.; Andes, D.; Brown, S.D.; Chaturvedi, V.; Ghannoum, M.A.; Knapp, C.C.; Sheehan, D.J.; Walsh, T.J. Correlation of MIC with Outcome for Candida Species Tested against Caspofungin, Anidulafungin, and Micafungin: Analysis and Proposal for Interpretive MIC Breakpoints. Journal of Clinical Microbiology 2008, 46(8), 2620-2629. 10.1128/jcm.00566-08.
- Kabir, V.; Maertens, J.; Kuypers, D. Fungal infections in solid organ transplantation: An update on diagnosis and treatment. Transplant. Rev. 2019, 33(2), 77-86. https://doi.org/10.1016/j.trre.2018.12.001.
- Hakim, A.; Braun, H.; Thornton, D.; Strymish, J. Successful treatment of methicillin-sensitive Staphylococcus aureus tricuspid-valve endocarditis with dalbavancin as an outpatient in a person who injects drugs: A case report. Int. J. Infect. Dis. 2020, 91, 202-205. https://doi.org/10.1016/j.ijid.2019.12.008.
- Chen, A.Y.; Zervos, M.J.; Vazquez, J.A. Dalbavancin: a novel antimicrobial. Int J Clin Pract 2007, 61(5), 853-863. 10.1111/j.1742-1241.2007.01318.x.
- Giannella, M.; Bartoletti, M.; Gatti, M.; Viale, P. Advances in the therapy of bacterial bloodstream infections. Clin. Microbiol. Infect. 2020, 26(2), 158-167. https://doi.org/10.1016/j.cmi.2019.11.001.
- Berditsch, M.; Lux, H.; Babii, O.; Afonin, S.; Ulrich, A.S. Therapeutic Potential of Gramicidin S in the Treatment of Root Canal Infections. Pharmaceuticals (Basel, Switzerland) 2016, 9(3). 10.3390/ph9030056.
- Mirski, T.; Niemcewicz, M.; Bartoszcze, M.; Gryko, R.; Michalski, A. Utilisation of peptides against microbial infections – a review. Annals of Agricultural and Environmental Medicine 2018, 25(2), 205-210. https://doi.org/10.26444/aaem/74471.
- Loutet, S.A.; Valvano, M.A. Extreme antimicrobial Peptide and polymyxin B resistance in the genus burkholderia. Front Microbiol 2011, 2, 159-159. 10.3389/fmicb.2011.00159.
- Gomes, B.; Augusto, M.T.; Felício, M.R.; Hollmann, A.; Franco, O.L.; Gonçalves, S.; Santos, N.C. Designing improved active peptides for therapeutic approaches against infectious diseases. Biotechnology Advances 2018, 36(2), 415-429. https://doi.org/10.1016/j.biotechadv.2018.01.004.
- Brade, K.D.; Rybak, J.M.; Rybak, M.J. Oritavancin: A New Lipoglycopeptide Antibiotic in the Treatment of Gram-Positive Infections. Infect Dis Ther 2016, 5(1), 1-15. 10.1007/s40121-016-0103-4.
- Kosowska-Shick, K.; Clark, C.; Pankuch, G.A.; Mcghee, P.; Dewasse, B.; Beachel, L.; Appelbaum, P.C. Activity of Telavancin against Staphylococci and Enterococci Determined by MIC and Resistance Selection Studies. Antimicrob Agents Chemother 2009, 53(10), 4217-4224. 10.1128/aac.00742-09.
- Leshem, A.; Liwinski, T.; Elinav, E. Immune-Microbiota Interplay and Colonization Resistance in Infection. Molecular Cell 2020. https://doi.org/10.1016/j.molcel.2020.03.001.
- Bauer, M.E.; Shafer, W.M. On the in vivo significance of bacterial resistance to antimicrobial peptides. Biochimica et Biophysica Acta (BBA) - Biomembranes 2015, 1848(11, Part B), 3101-3111. https://doi.org/10.1016/j.bbamem.2015.02.012.
- Lewies, A.; Du Plessis, L.H.; Wentzel, J.F. Antimicrobial Peptides: the Achilles’ Heel of Antibiotic Resistance? Probiotics and Antimicrobial Proteins 2019, 11(2), 370-381. https://doi.org/10.1007/s12602-018-9465-0.
- Kawano, Y.; Jordan, O.; Hanawa, T.; Borchard, G.; Patrulea, V. Are Antimicrobial Peptide Dendrimers an Escape from ESKAPE? Advances in wound care 2020, 9(7), 378–395. https://doi.org/10.1089/wound.2019.1113.
- Park, S.-C.; Park, Y.; Hahm, K.-S. The role of antimicrobial peptides in preventing multidrug-resistant bacterial infections and biofilm formation. Int J Mol Sci 2011, 12(9), 5971-5992. https://doi.org/10.3390/ijms12095971.
- Kumar, P.; Pletzer, D.; Haney, E.F.; Rahanjam, N.; Cheng, J.T.J.; Yue, M.; Aljehani, W.; Hancock, R.E.W.; Kizhakkedathu, J.N.; Straus, S.K. Aurein-Derived Antimicrobial Peptides Formulated with Pegylated Phospholipid Micelles to Target Methicillin-Resistant Staphylococcus aureus Skin Infections. ACS Infectious Diseases 2019, 5(3), 443-453. https://doi.org/10.1021/acsinfecdis.8b00319.
- Tabatabaei Mirakabad, F.S.; Khoramgah, M.S.; Keshavarz F, K.; Tabarzad, M.; Ranjbari, J. Peptide dendrimers as valuable biomaterials in medical sciences. Life sciences 2019, 233, 116754. 10.1016/j.lfs.2019.116754.
- Siriwardena, T.N.; Lüscher, A.; Köhler, T.; Van Delden, C.; Javor, S.; Reymond, J.-L. Antimicrobial Peptide Dendrimer Chimera. Helvetica Chimica Acta 2019, 102(4), e1900034. https://doi.org/10.1002/hlca.201900034.
- Tam, J.P.; Lu, Y.-A.; Yang, J.-L. Antimicrobial dendrimeric peptides. European Journal of Biochemistry 2002, 269(3), 923-932. https://doi.org/10.1046/j.0014-2956.2001.02728.x.
- Sowińska, M.; Laskowska, A.; Guśpiel, A.; Solecka, J.; Bochynska-Czyż, M.; Lipkowski, A.W.; Trzeciak, K.; Urbanczyk-Lipkowska, Z. Bioinspired Amphiphilic Peptide Dendrimers as Specific and Effective Compounds against Drug Resistant Clinical Isolates of E. coli. Bioconjugate Chemistry 2018, 29(11), 3571-3585. https://doi.org/10.1021/acs.bioconjchem.8b00544.
- Pires, J.; Siriwardena, T.N.; Stach, M.; Tinguely, R.; Kasraian, S.; Luzzaro, F.; Leib, S.L.; Darbre, T.; Reymond, J.L.; Endimiani, A. In Vitro Activity of the Novel Antimicrobial Peptide Dendrimer G3KL against Multidrug-Resistant Acinetobacter baumannii and Pseudomonas aeruginosa. Antimicrob Agents Chemother 2015, 59(12), 7915-7918. https://doi.org/10.1128/aac.01853-15.
- Han, X.; Liu, Y.; Ma, Y.; Zhang, M.; He, Z.; Siriwardena, T.N.; Xu, H.; Bai, Y.; Zhang, X.; Reymond, J.-L.; Qiao, M. Peptide dendrimers G3KL and TNS18 inhibit Pseudomonas aeruginosa biofilms. Applied Microbiology and Biotechnology 2019, 103(14), 5821-5830. https://doi.org/10.1007/s00253-019-09801-3.
- Gan, B.-H.; Siriwardena, T.N.; Javor, S.; Darbre, T.; Reymond, J.-L. Fluorescence Imaging of Bacterial Killing by Antimicrobial Peptide Dendrimer G3KL. ACS Infectious Diseases 2019, 5(12), 2164-2173. https://doi.org/10.1021/acsinfecdis.9b00299.
- Pompilio, A.; Geminiani, C.; Mantini, P.; Siriwardena, T.N.; Di Bonaventura, I.; Reymond, J.L.; Di Bonaventura, G. Peptide dendrimers as "lead compounds" for the treatment of chronic lung infections by Pseudomonas aeruginosa in cystic fibrosis patients: in vitro and in vivo studies. Infection and drug resistance 2018, 11, 1767-1782. https://doi.org/10.2147/idr.s168868.
- Scorciapino, M.A.; Serra, I.; Manzo, G.; Rinaldi, A.C. Antimicrobial Dendrimeric Peptides: Structure, Activity and New Therapeutic Applications. Int J Mol Sci 2017, 18(3). https://doi.org/10.3390/ijms18030542.
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: current status and future directions. Drug Discovery Today 2015, 20(1), 122-128. https://doi.org/10.1016/j.drudis.2014.10.003.
- Da Cunha, N.B.; Cobacho, N.B.; Viana, J.F.C.; Lima, L.A.; Sampaio, K.B.O.; Dohms, S.S.M.; Ferreira, A.C.R.; De La Fuente-Núñez, C.; Costa, F.F.; Franco, O.L.; Dias, S.C. The next generation of antimicrobial peptides (AMPs) as molecular therapeutic tools for the treatment of diseases with social and economic impacts. Drug Discovery Today 2017, 22(2), 234-248. https://doi.org/10.1016/j.drudis.2016.10.017.
- Laws, M.; Shaaban, A.; Rahman, K.M. Antibiotic resistance breakers: current approaches and future directions. FEMS Microbiology Reviews 2019, 43(5), 490-516. 10.1093/femsre/fuz014.
- González-Bello, C. Antibiotic adjuvants – A strategy to unlock bacterial resistance to antibiotics. Bioorganic & Medicinal Chemistry Letters 2017, 27(18), 4221-4228. https://doi.org/10.1016/j.bmcl.2017.08.027.
- Brown, D. Antibiotic resistance breakers: can repurposed drugs fill the antibiotic discovery void? Nature Reviews Drug Discovery 2015, 14(12), 821-832. 10.1038/nrd4675.
- Vasile, C.; Pamfil, D.; Stoleru, E.; Baican, M. New Developments in Medical Applications of Hybrid Hydrogels Containing Natural Polymers. Molecules 2020, 25(7). https://doi.org/10.3390/molecules25071539.
- Townsend, L.; Williams, R.L.; Anuforom, O.; Berwick, M.R.; Halstead, F.; Hughes, E.; Stamboulis, A.; Oppenheim, B.; Gough, J.; Grover, L.; Scott, R.a.H.; Webber, M.; Peacock, A.F.A.; Belli, A.; Logan, A.; Cogan, F.D. Antimicrobial peptide coatings for hydroxyapatite: electrostatic and covalent attachment of antimicrobial peptides to surfaces. Journal of The Royal Society Interface 2017, 14(126), 20160657. https://doi.org/doi:10.1098/rsif.2016.0657.
- Patrulea, V.; Hirt-Burri, N.; Jeannerat, A.; Applegate, L.A.; Ostafe, V.; Jordan, O.; Borchard, G. Peptide-decorated chitosan derivatives enhance fibroblast adhesion and proliferation in wound healing. Carbohydrate polymers 2016, 142, 114-123. https://doi.org/10.1016/j.carbpol.2016.01.045.
- Patrulea, V.; Laurent-Applegate, L.A.; Ostafe, V.; Borchard, G.; Jordan, O. Polyelectrolyte nanocomplexes based on chitosan derivatives for wound healing application. European Journal of Pharmaceutics and Biopharmaceutics 2019, 140, 100-108. https://doi.org/10.1016/j.ejpb.2019.05.009.
- Barbosa, M.; Costa, F.; Monteiro, C.; Duarte, F.; Martins, M.C.L.; Gomes, P. Antimicrobial coatings prepared from Dhvar-5-click-grafted chitosan powders. Acta Biomaterialia 2019, 84, 242-256. https://doi.org/10.1016/j.actbio.2018.12.001.
- Monteiro, C.; Fernandes, H.; Oliveira, D.; Vale, N.; Barbosa, M.; Gomes, P.; Mc, L.M. AMP-Chitosan Coating with Bactericidal Activity in the Presence of Human Plasma Proteins. Molecules 2020, 25(13), E3046. 10.3390/molecules25133046.
- Veronese, F.M.; Mero, A. The Impact of PEGylation on Biological Therapies. BioDrugs 2008, 22(5), 315-329. https://doi.org/10.2165/00063030-200822050-00004.
- Imura, Y.; Nishida, M.; Ogawa, Y.; Takakura, Y.; Matsuzaki, K. Action mechanism of tachyplesin I and effects of PEGylation. Biochimica et Biophysica Acta (BBA) - Biomembranes 2007, 1768(5), 1160-1169. https://doi.org/10.1016/j.bbamem.2007.01.005.
- Imura, Y.; Nishida, M.; Matsuzaki, K. Action mechanism of PEGylated magainin 2 analogue peptide. Biochimica et Biophysica Acta (BBA) - Biomembranes 2007, 1768(10), 2578-2585. https://doi.org/10.1016/j.bbamem.2007.06.013.
- Singh, S.; Papareddy, P.; Mörgelin, M.; Schmidtchen, A.; Malmsten, M. Effects of PEGylation on Membrane and Lipopolysaccharide Interactions of Host Defense Peptides. Biomacromolecules 2014, 15(4), 1337-1345. https://doi.org/10.1021/bm401884e.
- Papathanasiou, E.; Finkelman, M.; Hanley, J.; Parashis, A.O. Prevalence, Etiology and Treatment of Peri-Implant Mucositis and Peri-Implantitis: A Survey of Periodontists in the United States. J. Periodontol. 2016, 87(5), 493-501. https://doi.org/10.1902/jop.2015.150476.
- Dutta, D.; Ozkan, J.; Willcox, M.D. Biocompatibility of antimicrobial melimine lenses: rabbit and human studies. Optom. Vis. Sci. 2014, 91(5), 570-581. https://doi.org/10.1097/opx.0000000000000232.
- Dutta, D.; Zhao, T.; Cheah, K.B.; Holmlund, L.; Willcox, M.D.P. Activity of a melimine derived peptide Mel4 against Stenotrophomonas, Delftia, Elizabethkingia, Burkholderia and biocompatibility as a contact lens coating. Contact Lens Anterio. 2017, 40(3), 175-183. https://doi.org/10.1016/j.clae.2017.01.002.
- Taotao, F.; Xiaoyan, Y.; Bing, S.; Leming, S. Peptide self-assembled nanostructures for drug delivery applications. Journal of Nanomaterials 2017, 2017, 16. https://doi.org/10.1155/2017/4562474.
- Itzhaki, R.F.; Lathe, R.; Balin, B.J.; Ball, M.J.; Bearer, E.L.; Braak, H.; Bullido, M.J.; Carter, C.; Clerici, M.; Cosby, S.L.; Del Tredici, K.; Field, H.; Fulop, T.; Grassi, C.; Griffin, W.S.T.; Haas, J.; Hudson, A.P.; Kamer, A.R.; Kell, D.B.; Licastro, F.; Letenneur, L.; Lövheim, H.; Mancuso, R.; Miklossy, J.; Otth, C.; Palamara, A.T.; Perry, G.; Preston, C.; Pretorius, E.; Strandberg, T.; Tabet, N.; Taylor-Robinson, S.D.; Whittum-Hudson, J.A. Microbes and Alzheimer's Disease. Journal of Alzheimer's Disease 2016, 51(4), 979-984. https://doi.org/10.3233/JAD-160152.
- Bourdenx, M.; Koulakiotis, N.S.; Sanoudou, D.; Bezard, E.; Dehay, B.; Tsarbopoulos, A. Protein aggregation and neurodegeneration in prototypical neurodegenerative diseases: Examples of amyloidopathies, tauopathies and synucleinopathies. Progress in Neurobiology 2017, 155, 171-193. https://doi.org/10.1016/j.pneurobio.2015.07.003.
- Liu, Y.; Ren, B.; Zhang, Y.; Sun, Y.; Chang, Y.; Liang, G.; Xu, L.; Zheng, J. Molecular simulation aspects of amyloid peptides at membrane interface. Biochimica et Biophysica Acta (BBA) - Biomembranes 2018, 1860(9), 1906-1916. https://doi.org/10.1016/j.bbamem.2018.02.004.
- Malekkhaiat Häffner, S.; Malmsten, M. Influence of self-assembly on the performance of antimicrobial peptides. Current Opinion in Colloid & Interface Science 2018, 38, 56-79. https://doi.org/10.1016/j.cocis.2018.09.002.
- Görbitz, C.H. The structure of nanotubes formed by diphenylalanine, the core recognition motif of Alzheimer's β-amyloid polypeptide. Chemical Communications 2006, (22), 2332-2334. https://doi.org/10.1039/B603080G.
- Krysmann, M.J.; Castelletto, V.; Kelarakis, A.; Hamley, I.W.; Hule, R.A.; Pochan, D.J. Self-Assembly and Hydrogelation of an Amyloid Peptide Fragment. Biochemistry 2008, 47(16), 4597-4605. https://doi.org/10.1021/bi8000616.
- Last, N.B.; Miranker, A.D. Common mechanism unites membrane poration by amyloid and antimicrobial peptides. Proceedings of the National Academy of Sciences 2013, 110(16), 6382-6387. https://doi.org/10.1073/pnas.1219059110.
- Chen, W.; Yang, S.; Li, S.; Lang, J.C.; Mao, C.; Kroll, P.; Tang, L.; Dong, H. Self-Assembled Peptide Nanofibers Display Natural Antimicrobial Peptides to Selectively Kill Bacteria without Compromising Cytocompatibility. ACS Applied Materials & Interfaces 2019, 11(32), 28681-28689. https://doi.org/10.1021/acsami.9b09583.
- Yazici, H.; Habib, G.; Boone, K.; Urgen, M.; Utku, F.S.; Tamerler, C. Self-assembling antimicrobial peptides on nanotubular titanium surfaces coated with calcium phosphate for local therapy. Materials Science and Engineering: C 2019, 94, 333-343. https://doi.org/10.1016/j.msec.2018.09.030.
- Hong, W.; Zhao, Y.; Guo, Y.; Huang, C.; Qiu, P.; Zhu, J.; Chu, C.; Shi, H.; Liu, M. PEGylated Self-Assembled Nano-Bacitracin A: Probing the Antibacterial Mechanism and Real-Time Tracing of Target Delivery in Vivo. ACS Applied Materials & Interfaces 2018, 10(13), 10688-10705. https://doi.org/10.1021/acsami.8b00135.
- Cirioni, O.; Silvestri, C.; Ghiselli, R.; Orlando, F.; Riva, A.; Mocchegiani, F.; Chiodi, L.; Castelletti, S.; Gabrielli, E.; Saba, V.; Scalise, G.; Giacometti, A. Protective effects of the combination of α-helical antimicrobial peptides and rifampicin in three rat models of Pseudomonas aeruginosa infection. Journal of Antimicrobial Chemotherapy 2008, 62(6), 1332-1338. https://doi.org/10.1093/jac/dkn393.
- Cirioni, O.; Ghiselli, R.; Silvestri, C.; Kamysz, W.; Orlando, F.; Mocchegiani, F.; Di Matteo, F.; Riva, A.; Łukasiak, J.; Scalise, G.; Saba, V.; Giacometti, A. Efficacy of tachyplesin III, colistin, and imipenem against a multiresistant Pseudomonas aeruginosa strain. Antimicrob Agents Chemother 2007, 51(6), 2005. https://doi.org/10.1128/AAC.01576-06.
- Jeong, N.; Kim, J.-Y.; Park, S.-C.; Lee, J.-K.; Gopal, R.; Yoo, S.; Son, B.K.; Hahm, J.S.; Park, Y.; Hahm, K.-S. Antibiotic and synergistic effect of Leu–Lys rich peptide against antibiotic resistant microorganisms isolated from patients with cholelithiasis. Biochemical and Biophysical Research Communications 2010, 399(4), 581-586. https://doi.org/10.1016/j.bbrc.2010.07.118.
- Lakshminarayanan, R.; Tan, W.X.; Aung, T.T.; Goh, E.T.L.; Muruganantham, N.; Li, J.; Chang, J.Y.T.; Dikshit, N.; Saraswathi, P.; Lim, R.R.; Kang, T.S.; Balamuralidhar, V.; Sukumaran, B.; Verma, C.S.; Sivaraman, J.; Chaurasia, S.S.; Liu, S.; Beuerman, R.W. Branched Peptide, B2088, Disrupts the Supramolecular Organization of Lipopolysaccharides and Sensitizes the Gram-negative Bacteria. Sci Rep 2016, 6(1), 25905. https://doi.org/10.1038/srep25905.
- Dosler, S.; Karaaslan, E. Inhibition and destruction of Pseudomonas aeruginosa biofilms by antibiotics and antimicrobial peptides. Peptides 2014, 62, 32-37. https://doi.org/10.1016/j.peptides.2014.09.021.
- Chen, H.; Wubbolts, R.W.; Haagsman, H.P.; Veldhuizen, E.J.A. Inhibition and Eradication of Pseudomonas aeruginosa Biofilms by Host Defence Peptides. Sci Rep 2018, 8(1), 10446-10446. https://doi.org/10.1038/s41598-018-28842-8.
- Tyers, M.; Wright, G.D. Drug combinations: a strategy to extend the life of antibiotics in the 21st century. Nature Reviews Microbiology 2019, 17(3), 141-155. https://doi.org/10.1038/s41579-018-0141-x.
- Pal, I.; Bhattacharyya, D.; Kar, R.K.; Zarena, D.; Bhunia, A.; Atreya, H.S. A Peptide-Nanoparticle System with Improved Efficacy against Multidrug Resistant Bacteria. Sci Rep 2019, 9(1), 4485. https://doi.org/10.1038/s41598-019-41005-7.
- Bahrami, A.; Delshadi, R.; Jafari, S.M. Active delivery of antimicrobial nanoparticles into microbial cells through surface functionalization strategies. Trends in Food Science & Technology 2020, 99, 217-228. https://doi.org/10.1016/j.tifs.2020.03.008.
- Borro, B.C.; Malmsten, M. Complexation between antimicrobial peptides and polyelectrolytes. Advances in Colloid and Interface Science 2019, 270, 251-260. https://doi.org/10.1016/j.cis.2019.07.001.
- Nikolova, M.P.; Chavali, M.S. Recent advances in biomaterials for 3D scaffolds: A review. Bioact Mater 2019, 4, 271-292. https://doi.org/10.1016/j.bioactmat.2019.10.005.
- Calori, I.R.; Braga, G.; Carvalho De Jesus, P.D.C.; Bi, H.; Tedesco, A.C. Polymer Scaffolds as Drug Delivery Systems. European Polymer Journal 2020, 109621. https://doi.org/10.1016/j.eurpolymj.2020.109621.
- Liu, C.; Zhang, Q.; Zhu, S.; Liu, H.; Chen, J. Preparation and applications of peptide-based injectable hydrogels. RSC Advances 2019, 9(48), 28299-28311. https://doi.org/10.1039/C9RA05934B.
- Lozeau, L.D.; Grosha, J.; Kole, D.; Prifti, F.; Dominko, T.; Camesano, T.A.; Rolle, M.W. Collagen tethering of synthetic human antimicrobial peptides cathelicidin LL37 and its effects on antimicrobial activity and cytotoxicity. Acta Biomaterialia 2017, 52, 9-20. https://doi.org/10.1016/j.actbio.2016.12.047.
- Cassin, M.E.; Ford, A.J.; Orbach, S.M.; Saverot, S.E.; Rajagopalan, P. The design of antimicrobial LL37-modified collagen-hyaluronic acid detachable multilayers. Acta Biomaterialia 2016, 40, 119-129. https://doi.org/10.1016/j.actbio.2016.04.027.
- US National Library of Medicine. A Study of Granexin Gel in the Treatment of Diabetic Foot Ulcer https://clinicaltrials.gov/ct2/show/NCT02667327 (accessed 23/03/2020)
- Grek, C.L.; Prasad, G.M.; Viswanathan, V.; Armstrong, D.G.; Gourdie, R.G.; Ghatnekar, G.S. Topical administration of a connexin43-based peptide augments healing of chronic neuropathic diabetic foot ulcers: A multicenter, randomized trial. Wound Repair Regen 2015, 23(2), 203-212. https://doi.org/10.1111/wrr.12275.
- Sun, T.; Zhan, B.; Zhang, W.; Qin, D.; Xia, G.; Zhang, H.; Peng, M.; Li, S.-A.; Zhang, Y.; Gao, Y.; Lee, W.-H. Carboxymethyl chitosan nanoparticles loaded with bioactive peptide OH-CATH30 benefit nonscar wound healing. International journal of nanomedicine 2018, 13, 5771-5786. https://doi.org/10.2147/IJN.S156206.
- Almaaytah, A.; Mohammed, G.K.; Abualhaijaa, A.; Al-Balas, Q. Development of novel ultrashort antimicrobial peptide nanoparticles with potent antimicrobial and antibiofilm activities against multidrug-resistant bacteria. Drug design, development and therapy 2017, 11, 3159-3170. https://doi.org/10.2147/DDDT.S147450.
- Nordström, R.; Nyström, L.; Andrén, O.C.J.; Malkoch, M.; Umerska, A.; Davoudi, M.; Schmidtchen, A.; Malmsten, M. Membrane interactions of microgels as carriers of antimicrobial peptides. Journal of Colloid and Interface Science 2018, 513, 141-150. https://doi.org/10.1016/j.jcis.2017.11.014.
- Nordström, R.; Browning, K.L.; Parra-Ortiz, E.; Damgaard, L.S.E.; Häffner, S.M.; Maestro, A.; Campbell, R.A.; Cooper, J.F.K.; Malmsten, M. Membrane interactions of antimicrobial peptide-loaded microgels. J. Colloid Interface Sci. 2020, 562, 322-332. https://doi.org/10.1016/j.jcis.2019.12.022.
- Boge, L.; Hallstensson, K.; Ringstad, L.; Johansson, J.; Andersson, T.; Davoudi, M.; Larsson, P.T.; Mahlapuu, M.; Håkansson, J.; Andersson, M. Cubosomes for topical delivery of the antimicrobial peptide LL-37. European Journal of Pharmaceutics and Biopharmaceutics 2019, 134, 60-67. https://doi.org/10.1016/j.ejpb.2018.11.009.
- Innocenti Malini, R.; Zabara, M.; Gontsarik, M.; Maniura-Weber, K.; Rossi, R.M.; Spano, F.; Salentinig, S. Self-assembly of glycerol monooleate with the antimicrobial peptide LL-37: a molecular dynamics study. RSC Advances 2020, 10(14), 8291-8302. https://doi.org/10.1039/C9RA10037G.
- Silva, J.P.; Gonçalves, C.; Costa, C.; Sousa, J.; Silva-Gomes, R.; Castro, A.G.; Pedrosa, J.; Appelberg, R.; Gama, F.M. Delivery of LLKKK18 loaded into self-assembling hyaluronic acid nanogel for tuberculosis treatment. Journal of Controlled Release 2016, 235, 112-124. https://doi.org/10.1016/j.jconrel.2016.05.064.
- Lin, Z.; Wu, T.; Wang, W.; Li, B.; Wang, M.; Chen, L.; Xia, H.; Zhang, T. Biofunctions of antimicrobial peptide-conjugated alginate/hyaluronic acid/collagen wound dressings promote wound healing of a mixed-bacteria-infected wound. International Journal of Biological Macromolecules 2019, 140, 330-342. https://doi.org/10.1016/j.ijbiomac.2019.08.087.
- Chereddy, K.K.; Her, C.-H.; Comune, M.; Moia, C.; Lopes, A.; Porporato, P.E.; Vanacker, J.; Lam, M.C.; Steinstraesser, L.; Sonveaux, P.; Zhu, H.; Ferreira, L.S.; Vandermeulen, G.; Préat, V. PLGA nanoparticles loaded with host defense peptide LL37 promote wound healing. Journal of Controlled Release 2014, 194, 138-147. https://doi.org/10.1016/j.jconrel.2014.08.016.
- Casciaro, B.; D’angelo, I.; Zhang, X.; Loffredo, M.R.; Conte, G.; Cappiello, F.; Quaglia, F.; Di, Y.-P.P.; Ungaro, F.; Mangoni, M.L. Poly(lactide-co-glycolide) Nanoparticles for Prolonged Therapeutic Efficacy of Esculentin-1a-Derived Antimicrobial Peptides against Pseudomonas aeruginosa Lung Infection: in Vitro and in Vivo Studies. Biomacromolecules 2019, 20(5), 1876-1888. https://doi.org/10.1021/acs.biomac.8b01829.
- Wang, C.; Feng, S.; Qie, J.; Wei, X.; Yan, H.; Liu, K. Polyion complexes of a cationic antimicrobial peptide as a potential systemically administered antibiotic. International Journal of Pharmaceutics 2019, 554, 284-291. https://doi.org/10.1016/j.ijpharm.2018.11.029.
- Garcia-Orue, I.; Gainza, G.; Girbau, C.; Alonso, R.; Aguirre, J.J.; Pedraz, J.L.; Igartua, M.; Hernandez, R.M. LL37 loaded nanostructured lipid carriers (NLC): A new strategy for the topical treatment of chronic wounds. European Journal of Pharmaceutics and Biopharmaceutics 2016, 108, 310-316. https://doi.org/10.1016/j.ejpb.2016.04.006.
- Cheng, R.; Liu, L.; Xiang, Y.; Lu, Y.; Deng, L.; Zhang, H.; Santos, H.A.; Cui, W. Advanced liposome-loaded scaffolds for therapeutic and tissue engineering applications. Biomaterials 2020, 232, 119706. https://doi.org/10.1016/j.biomaterials.2019.119706.
- Wijnant, G.-J.; Van Bocxlaer, K.; Yardley, V.; Harris, A.; Alavijeh, M.; Silva-Pedrosa, R.; Antunes, S.; Mauricio, I.; Murdan, S.; Croft, S.L. Comparative efficacy, toxicity and biodistribution of the liposomal amphotericin B formulations Fungisome® and AmBisome® in murine cutaneous leishmaniasis. International Journal for Parasitology: Drugs and Drug Resistance 2018, 8(2), 223-228. https://doi.org/10.1016/j.ijpddr.2018.04.001.
- Yang, Z.; Liu, J.; Gao, J.; Chen, S.; Huang, G. Chitosan coated vancomycin hydrochloride liposomes: Characterizations and evaluation. International Journal of Pharmaceutics 2015, 495(1), 508-515. https://doi.org/10.1016/j.ijpharm.2015.08.085.
- Yamakami, K.; Tsumori, H.; Shimizu, Y.; Sakurai, Y.; Nagatoshi, K.; Sonomoto, K. Cationic Lipid Content in Liposome-Encapsulated Nisin Improves Sustainable Bactericidal Activity against Streptococcus mutans. Open Dent J 2016, 10, 360-366. https://doi.org/10.2174/1874210616021001360.
- Alipour, M.; Suntres, Z.E.; Halwani, M.; Azghani, A.O.; Omri, A. Activity and interactions of liposomal antibiotics in presence of polyanions and sputum of patients with cystic fibrosis. PloS one 2009, 4(5), e5724. https://doi.org/10.1371/journal.pone.0005724.
- He, J.; Abdelraouf, K.; Ledesma, K.R.; Chow, D.S.L.; Tam, V.H. Pharmacokinetics and efficacy of liposomal polymyxin B in a murine pneumonia model. International Journal of Antimicrobial Agents 2013, 42(6), 559-564. https://doi.org/10.1016/j.ijantimicag.2013.07.009.
- Li, C.; Zhang, X.; Huang, X.; Wang, X.; Liao, G.; Chen, Z. Preparation and characterization of flexible nanoliposomes loaded with daptomycin, a novel antibiotic, for topical skin therapy. International journal of nanomedicine 2013, 8, 1285-1292. https://doi.org/10.2147/IJN.S41695.
- Prasher, P.; Sharma, M.; Mudila, H.; Gupta, G.; Sharma, A.K.; Kumar, D.; Bakshi, H.A.; Negi, P.; Kapoor, D.N.; Chellappan, D.K.; Tambuwala, M.M.; Dua, K. Emerging trends in clinical implications of bio-conjugated silver nanoparticles in drug delivery. Colloid and Interface Science Communications 2020, 35, 100244. https://doi.org/10.1016/j.colcom.2020.100244.
- Casciaro, B.; Moros, M.; Rivera-Fernández, S.; Bellelli, A.; De La Fuente, J.M.; Mangoni, M.L. Gold-nanoparticles coated with the antimicrobial peptide esculentin-1a(1-21)NH2 as a reliable strategy for antipseudomonal drugs. Acta Biomaterialia 2017, 47, 170-181. https://doi.org/10.1016/j.actbio.2016.09.041.
- De Alteriis, E.; Maselli, V.; Falanga, A.; Galdiero, S.; Di Lella, F.M.; Gesuele, R.; Guida, M.; Galdiero, E. Efficiency of gold nanoparticles coated with the antimicrobial peptide indolicidin against biofilm formation and development of Candida spp. clinical isolates. Infection and drug resistance 2018, 11, 915-925. https://doi.org/10.2147/idr.s164262.
- Rai, A.; Pinto, S.; Velho, T.R.; Ferreira, A.F.; Moita, C.; Trivedi, U.; Evangelista, M.; Comune, M.; Rumbaugh, K.P.; Simões, P.N.; Moita, L.; Ferreira, L. One-step synthesis of high-density peptide-conjugated gold nanoparticles with antimicrobial efficacy in a systemic infection model. Biomaterials 2016, 85, 99-110. https://doi.org/10.1016/j.biomaterials.2016.01.051.
- Pal, I.; Brahmkhatri, V.P.; Bera, S.; Bhattacharyya, D.; Quirishi, Y.; Bhunia, A.; Atreya, H.S. Enhanced stability and activity of an antimicrobial peptide in conjugation with silver nanoparticle. Journal of Colloid and Interface Science 2016, 483, 385-393. https://doi.org/10.1016/j.jcis.2016.08.043.
- Lambadi, P.R.; Sharma, T.K.; Kumar, P.; Vasnani, P.; Thalluri, S.M.; Bisht, N.; Pathania, R.; Navani, N.K. Facile biofunctionalization of silver nanoparticles for enhanced antibacterial properties, endotoxin removal, and biofilm control. International journal of nanomedicine 2015, 10, 2155-2171. https://doi.org/10.2147/ijn.s72923.
- Zheng, K.; Setyawati, M.I.; Lim, T.-P.; Leong, D.T.; Xie, J. Antimicrobial Cluster Bombs: Silver Nanoclusters Packed with Daptomycin. ACS Nano 2016, 10(8), 7934-7942. https://doi.org/10.1021/acsnano.6b03862.
- Arvizo, R.; Bhattacharya, R.; Mukherjee, P. Gold nanoparticles: opportunities and challenges in nanomedicine. Expert Opin Drug Deliv 2010, 7(6), 753-763. https://doi.org/10.1517/17425241003777010.
- Balfourier, A.; Luciani, N.; Wang, G.; Lelong, G.; Ersen, O.; Khelfa, A.; Alloyeau, D.; Gazeau, F.; Carn, F. Unexpected intracellular biodegradation and recrystallization of gold nanoparticles. Proceedings of the National Academy of Sciences 2020, 117(1), 103-113. https://doi.org/10.1073/pnas.1911734116.
- Li, P.; Han, F.; Cao, W.; Zhang, G.; Li, J.; Zhou, J.; Gong, X.; Turnbull, G.; Shu, W.; Xia, L.; Fang, B.; Xing, X.; Li, B. Carbon quantum dots derived from lysine and arginine simultaneously scavenge bacteria and promote tissue repair. Applied Materials Today 2020, 19, 100601. https://doi.org/10.1016/j.apmt.2020.100601.
- Maleki Dizaj, S.; Mennati, A.; Jafari, S.; Khezri, K.; Adibkia, K. Antimicrobial Activity of Carbon-Based Nanoparticles. Adv Pharm Bull 2015, 5(1), 19-23. https://doi.org/10.5681/apb.2015.003.
- Chaudhari, A.A.; Ashmore, D.A.; Nath, S.D.; Kate, K.; Dennis, V.; Singh, S.R.; Owen, D.R.; Palazzo, C.; Arnold, R.D.; Miller, M.E.; Pillai, S.R. A novel covalent approach to bio-conjugate silver coated single walled carbon nanotubes with antimicrobial peptide. Journal of Nanobiotechnology 2016, 14(1), 58. https://doi.org/10.1186/s12951-016-0211-z.
- Sur, A.; Pradhan, B.; Banerjee, A.; Aich, P. Immune activation efficacy of indolicidin is enhanced upon conjugation with carbon nanotubes and gold nanoparticles. PloS one 2015, 10(4), e0123905-e0123905. https://doi.org/10.1371/journal.pone.0123905.
- Kanchanapally, R.; Viraka Nellore, B.P.; Sinha, S.S.; Pedraza, F.; Jones, S.J.; Pramanik, A.; Chavva, S.R.; Tchounwou, C.; Shi, Y.; Vangara, A.; Sardar, D.; Ray, P.C. Antimicrobial peptide-conjugated graphene oxide membrane for efficient removal and effective killing of multiple drug resistant bacteria. RSC Advances 2015, 5(24), 18881-18887. https://doi.org/10.1039/C5RA01321F.
- Viraka Nellore, B.P.; Kanchanapally, R.; Pedraza, F.; Sinha, S.S.; Pramanik, A.; Hamme, A.T.; Arslan, Z.; Sardar, D.; Ray, P.C. Bio-Conjugated CNT-Bridged 3D Porous Graphene Oxide Membrane for Highly Efficient Disinfection of Pathogenic Bacteria and Removal of Toxic Metals from Water. ACS Applied Materials & Interfaces 2015, 7(34), 19210-19218. https://doi.org/10.1021/acsami.5b05012.
- Dostalova, S.; Moulick, A.; Milosavljevic, V.; Guran, R.; Kominkova, M.; Cihalova, K.; Heger, Z.; Blazkova, L.; Kopel, P.; Hynek, D.; Vaculovicova, M.; Adam, V.; Kizek, R. Antiviral activity of fullerene C60 nanocrystals modified with derivatives of anionic antimicrobial peptide maximin H5. Monatshefte für Chemie - Chemical Monthly 2016, 147(5), 905-918. https://doi.org/10.1007/s00706-016-1675-0.
- Barron, A.R. [60]Fullerene-peptides: bio-nano conjugates with structural and chemical diversity. Journal of Enzyme Inhibition and Medicinal Chemistry 2016, 31(sup1), 164-176. https://doi.org/10.1080/14756366.2016.1177524.
- Pellarini, F.; Pantarotto, D.; Da Ros, T.; Giangaspero, A.; Tossi, A.; Prato, M. A Novel [60]Fullerene Amino Acid for Use in Solid-Phase Peptide Synthesis. Organic Letters 2001, 3(12), 1845-1848. https://doi.org/10.1021/ol015934m.
- Pantarotto, D.; Bianco, A.; Pellarini, F.; Tossi, A.; Giangaspero, A.; Zelezetsky, I.; Briand, J.-P.; Prato, M. Solid-Phase Synthesis of Fullerene-peptides. Journal of the American Chemical Society 2002, 124(42), 12543-12549. https://doi.org/10.1021/ja027603q.
- Ashraf, M.U.; Hussain, M.A.; Muhammad, G.; Haseeb, M.T.; Bashir, S.; Hussain, S.Z.; Hussain, I. A superporous and superabsorbent glucuronoxylan hydrogel from quince (Cydonia oblanga): Stimuli responsive swelling, on-off switching and drug release. International Journal of Biological Macromolecules 2017, 95, 138-144. https://doi.org/10.1016/j.ijbiomac.2016.11.057.
- Rivero, G.; Meuter, M.; Pepe, A.; Guevara, M.G.; Boccaccini, A.R.; Abraham, G.A. Nanofibrous membranes as smart wound dressings that release antibiotics when an injury is infected. Colloids and Surfaces A: Physicochemical and Engineering Aspects 2020, 587, 124313. https://doi.org/10.1016/j.colsurfa.2019.124313.
- Muhammad, G.; Hussain, M.A.; Amin, M.; Hussain, S.Z.; Hussain, I.; Abbas Bukhari, S.N.; Naeem-Ul-Hassan, M. Glucuronoxylan-mediated silver nanoparticles: green synthesis, antimicrobial and wound healing applications. RSC Advances 2017, 7(68), 42900-42908. https://doi.org/10.1039/C7RA07555C.
- Wu, K.; Wu, X.; Chen, M.; Wu, H.; Jiao, Y.; Zhou, C. H2O2-responsive smart dressing for visible H2O2 monitoring and accelerating wound healing. Chemical Engineering Journal 2020, 387, 124127. https://doi.org/10.1016/j.cej.2020.124127.
- Hu, M.; Korschelt, K.; Daniel, P.; Landfester, K.; Tremel, W.; Bannwarth, M.B. Fibrous Nanozyme Dressings with Catalase-Like Activity for H2O2 Reduction To Promote Wound Healing. ACS Applied Materials & Interfaces 2017, 9(43), 38024-38031. https://doi.org/10.1021/acsami.7b12212.
- Rezaei, N.; Hamidabadi, H.G.; Khosravimelal, S.; Zahiri, M.; Ahovan, Z.A.; Bojnordi, M.N.; Eftekhari, B.S.; Hashemi, A.; Ganji, F.; Darabi, S.; Gholipourmalekabadi, M. Antimicrobial peptides-loaded smart chitosan hydrogel: Release behavior and antibacterial potential against antibiotic resistant clinical isolates. International Journal of Biological Macromolecules 2020, 164, 855-862. https://doi.org/10.1016/j.ijbiomac.2020.07.011.
- Panzarasa, G.; Osypova, A.; Toncelli, C.; Buhmann, M.T.; Rottmar, M.; Ren, Q.; Maniura-Weber, K.; Rossi, R.M.; Boesel, L.F. The pyranine-benzalkonium ion pair: A promising fluorescent system for the ratiometric detection of wound pH. Sensors and Actuators B: Chemical 2017, 249, 156-160. https://doi.org/10.1016/j.snb.2017.04.045.
- Lombardi, L.; Shi, Y.; Falanga, A.; Galdiero, E.; De Alteriis, E.; Franci, G.; Chourpa, I.; Azevedo, H.S.; Galdiero, S. Enhancing the Potency of Antimicrobial Peptides through Molecular Engineering and Self-Assembly. Biomacromolecules 2019, 20(3), 1362-1374. https://doi.org/10.1021/acs.biomac.8b01740.
- Dimatteo, R.; Darling, N.J.; Segura, T. In situ forming injectable hydrogels for drug delivery and wound repair. Advanced drug delivery reviews 2018, 127, 167-184. https://doi.org/10.1016/j.addr.2018.03.007.
- Bradshaw, M.; Ho, D.; Fear, M.W.; Gelain, F.; Wood, F.M.; Iyer, K.S. Designer self-assembling hydrogel scaffolds can impact skin cell proliferation and migration. Sci Rep 2014, 4(1), 6903. 10.1038/srep06903.
- Zhou, A.; Chen, S.; He, B.; Zhao, W.; Chen, X.; Jiang, D. Controlled release of TGF-beta 1 from RADA self-assembling peptide hydrogel scaffolds. Drug design, development and therapy 2016, 10, 3043-3051. https://doi.org/10.2147/DDDT.S109545.
- Håkansson, J.; Ringstad, L.; Umerska, A.; Johansson, J.; Andersson, T.; Boge, L.; Rozenbaum, R.T.; Sharma, P.K.; Tollbäck, P.; Björn, C.; Saulnier, P.; Mahlapuu, M. Characterization of the in vitro, ex vivo, and in vivo Efficacy of the Antimicrobial Peptide DPK-060 Used for Topical Treatment. Frontiers in cellular and infection microbiology 2019, 9, 174-174. https://doi.org/10.3389/fcimb.2019.00174.
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic therapeutic peptides: science and market. Drug Discovery Today 2010, 15(1), 40-56. https://doi.org/10.1016/j.drudis.2009.10.009.
- Yamazaki, S.; Suzuki, T.; Suzuki, T.; Takatsuka, H.; Ishikawa, M.; Hattori, N.; Fujishiro, T.; Miyauchi, H.; Oami, T.; Ariyoshi, N.; Oda, S.; Matsubara, H.; Ishii, I. An extremely high bioavailability of orally administered vancomycin in a patient with severe colitis and renal insufficiency. Journal of Infection and Chemotherapy 2017, 23(12), 848-851. https://doi.org/10.1016/j.jiac.2017.08.004.
- Cimolai, N. Does oral vancomycin use necessitate therapeutic drug monitoring? Infection 2020, 48(2), 173-182. https://doi.org/10.1007/s15010-019-01374-7.
- Davis, C.A.; Janssen, E.M.L. Environmental fate processes of antimicrobial peptides daptomycin, bacitracins, and polymyxins. Environment International 2020, 134, 105271. https://doi.org/10.1016/j.envint.2019.105271.
- Saravolatz, L.D.; Cleveland, K.O.; Rikabi, K.; Hassoun, A.; Reilly, J.; Johnson, L.B.; Spak, C.; Valenti, S.; Szpunar, S. Real-world use of telavancin in the treatment of osteomyelitis. Diagnostic Microbiology and Infectious Disease 2019, 95(2), 185-190. https://doi.org/10.1016/j.diagmicrobio.2019.05.011.
- Chastain, D.B.; Davis, A. Treatment of chronic osteomyelitis with multidose oritavancin: A case series and literature review. International Journal of Antimicrobial Agents 2019, 53(4), 429-434. https://doi.org/10.1016/j.ijantimicag.2018.11.023.
- Sinha, R.; Shukla, P. Antimicrobial Peptides: Recent Insights on Biotechnological Interventions and Future Perspectives. Protein Pept. Lett. 2019, 26(2), 79-87. https://doi.org/10.2174/0929866525666181026160852.
- Pursey, E.; Sünderhauf, D.; Gaze, W.H.; Westra, E.R.; Van Houte, S. CRISPR-Cas antimicrobials: Challenges and future prospects. PLoS Pathog. 2018, 14 (6), e1006990. https://doi.org/10.1371/journal.ppat.1006990.
- Hanson, M.A.; Dostálová, A.; Ceroni, C.; Poidevin, M.; Kondo, S.; Lemaître, B. Correction: Synergy and remarkable specificity of antimicrobial peptides in vivo using a systematic knockout approach. eLife 2019, 8, e48778. https://doi.org/10.7554/eLife.44341.
- Unckless, Robert l.; Howick, Virginia m.; Lazzaro, Brian p. Convergent Balancing Selection on an Antimicrobial Peptide in Drosophila. Curr. Biol. 2016, 26(2), 257-262. https://doi.org/10.1016/j.cub.2015.11.063.
- Choudhary, E.; Sharma, R.; Kumar, Y.; Agarwal, N. Conditional Silencing by CRISPRi Reveals the Role of DNA Gyrase in Formation of Drug-Tolerant Persister Population in Mycobacterium tuberculosis. Front Cell Infect Microbiol 2019, 9, 70. https://doi.org/10.3389/fcimb.2019.00070.
- Wei, W.; Zhang, S.; Fleming, J.; Chen, Y.; Li, Z.; Fan, S.; Liu, Y.; Wang, W.; Wang, T.; Liu, Y.; Ren, B.; Wang, M.; Jiao, J.; Chen, Y.; Zhou, Y.; Zhou, Y.; Gu, S.; Zhang, X.; Wan, L.; Chen, T.; Zhou, L.; Chen, Y.; Zhang, X.E.; Li, C.; Zhang, H.; Bi, L. Mycobacterium tuberculosis type III-A CRISPR/Cas system crRNA and its maturation have atypical features. FASEB Journal 2019, 33(1), 1496-1509. https://doi.org/10.1096/fj.201800557RR.
- Citorik, R.J.; Mimee, M.; Lu, T.K. Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nat. Biotechnol. 2014, 32(11), 1141-1145. https://doi.org/10.1038/nbt.3011.
- Bikard, D.; Euler, C.W.; Jiang, W.; Nussenzweig, P.M.; Goldberg, G.W.; Duportet, X.; Fischetti, V.A.; Marraffini, L.A. Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat. Biotechnol. 2014, 32(11), 1146-1150. https://doi.org/10.1038/nbt.3043.
- Li, Y.; Peng, N. Endogenous CRISPR-Cas System-Based Genome Editing and Antimicrobials: Review and Prospects. Front Microbiol 2019, 10, 2471-2471. https://doi.org/10.3389/fmicb.2019.02471.
- Kosikowska, P.; Lesner, A. Antimicrobial peptides (AMPs) as drug candidates: a patent review (2003–2015). Expert Opinion on Therapeutic Patents 2016, 26(6), 689-702. https://doi.org/10.1080/13543776.2016.1176149.


