1000/1000
Hot
Most Recent
 +1 point
+1 point
The treatment of metastatic renal cell carcinoma has evolved quickly over the last few years from a disease managed primarily with sequential oral tyrosine kinase inhibitors (TKIs) targeting the vascular endothelial growth factor (VEGF) pathway, to now with a combination of therapies incorporating immune checkpoint blockade (ICB). Patient outcomes have improved with these innovations, however, controversy persists regarding the optimal sequence and patient selection amongst the available combinations. Ideally, predictive biomarkers would aid in guiding treatment decisions and personalizing care.
Renal cell carcinoma (RCC) is traditionally classified according to its histology. Clear cell (ccRCC) is the most common subtype, accounting for 75–85% of all RCCs. Current first-line standard of care therapies for metastatic ccRCC involve the use of vascular endothelial growth factor (VEGF) inhibitors, checkpoint inhibitors, anti-CTLA4 agents, or a combination of these drugs. Choice of therapy is guided by whether the patient’s disease falls under favorite or intermediate/poor risk based on validated prognostic models. Within each risk category, there are several acceptable alternatives, including VEGF inhibitor monotherapy, combination immunotherapy (e.g., ipilimumab/nivolumab), or a combination of a VEGF inhibitor and a checkpoint inhibitor (e.g., axitinib/pembrolizumab). Given the increasing number of available treatment options for mRCC there is also a growing need for predictive biomarkers to help guide clinicians (Figure 1). We review the literature regarding the evidence for selecting one type of regimen over another and determining who would benefit more from either angiogenesis antagonism or immune checkpoint blockade (ICB).
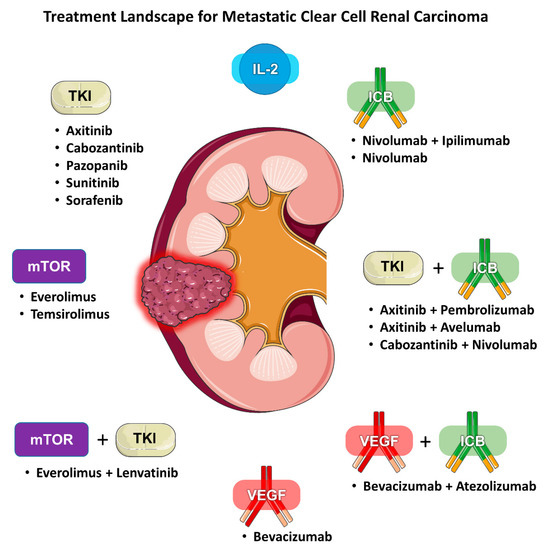
Figure 1. Treatment landscape for metastatic clear cell renal carcinoma.
Renal cell carcinoma is often considered an immunogenic tumor. This has been evidenced from pathologic examination of RCC tumor tissue showing significant infiltration by both T-cells and natural killer cells [1]. In addition, the efficacy of early immunotherapy agents, like interleukin-2 (IL-2) and Interferon-alpha (IFN-α), and more recently ICB in the treatment of RCC support this notion pragmatically [2][3][4]. ICB targeting the programmed death 1 (PD-1) and cytotoxic T-lymphocyte associated protein 4 (CTLA-4) pathways have demonstrated favorable outcomes, with ORRs of 25% for anti-PD-1 targeted single-agent therapy and up to 39% and 59% when combined with CTLA-4 or vascular endothelial growth factor (VEGF) inhibitors, respectively [5][6][7]. Consequently, combination strategies with ICB have become the standard of care for most eligible mRCC patients.
However, since both combination ICB/ICB and ICB/TKI regimens are approved as first-line therapy for mRCC, it would be beneficial to have clinical biomarkers to understand which tumors are more likely to benefit from an immunotherapy-based regimen versus a combination regimen with VEGF inhibition.
The programmed death-ligand 1 (PD-L1), also known as B7 homolog 1 (B7-H1), is found on tumor and immune cells in the TME, and its receptor PD-1 on T-cells are the primary targets for this form of ICB. In the era of ICB, expression of PD-L1 by immunohistochemistry (IHC) has been a focus of much biomarker research across tumor types but in the case of mRCC it has not borne out to be a very useful predictive biomarker.
When focusing specifically on registration studies for ICB in mRCC, patients without any measurable PD-L1 expression have benefited from these drugs. In a meta-analysis of six randomized controlled trials of ICB in mRCC an association was observed between PD-L1 expression and PFS, but the analysis failed to show a significant correlation with OS [8]. The authors concluded from this data that the role of PD-L1 expression in selecting treatment for RCC was not well established, in line with FDA drug approvals and the NCCN guidelines which do not include or require PD-L1 expression [9][10]. This difference is likely multifactorial and could be due to the unique biology of RCC, related to the non-standardized testing utilized in for PD-L1 expression as a biomarker in earlier trials, including the use of different antibodies for various IHC assays and inconsistent cutoffs for positivity, tumor heterogeneity and the dynamic nature of PD-L1 expression on tumor cells [11].
Furthermore, prior to the era of immunotherapy, PD-L1 expression by IHC was studied in mRCC and was shown to be associated with poor prognosis [12]. The observation that PD-L1 positivity is linked to poor prognosis was again reported more recently in a post-hoc analysis of the COMPARZ trial (pazopinib vs. sunitinib) which showed that patients who were PD-L1 positive had significantly worse OS and PFS compared to the PD-L1 negative population. This is also supported by an analysis of CHECKMATE-214 study (nivolumab+ipilumimab vs. sunitinib) which demonstrated that PD-L1 positivity was more common in patients with intermediate and poor-risk disease as defined by IMDC criteria compared to those with favorable-risk disease [6]. It is possible that the prognostic implications of PD-L1 positivity in mRCC also have a negative impact on its usefulness as a predictive biomarker.
Differences in the genomic landscape of RCC have also been the subject of much study in the search for clinical biomarkers for ICB treatment PBRM1 and PBAF complex mutations have drawn much attention in this regard and, as discussed above, have also been investigated as both a prognostic and predictive markers for VEGF TKIs. In relation to ICB, PBRM1 was first identified by Miao et al. in a set of 35 patients with mRCC who participated in a prospective clinical study of nivolumab. Whole-exome sequencing was performed on tissue samples and identified PBRM1 as being strongly enriched in the group that derived clinical benefit. This finding was then validated in a separate 63 patient cohort treated with PD-1 or PD-L1 inhibitions alone or in combination with anti-CTLA-4 therapies and replicated findings of association with clinical benefit [13].
However, after this initial publication, PBRM1 mutations were subsequently studied in several additional patient cohorts. An analysis by McDermott et al. of a first-line clinical trial of atezolizumab alone or in combination with bevacizumab vs. sunitinib failed to demonstrate an association with clinical benefit in patients with PBRM1 mutations in the atezolizumab monotherapy arm but instead favored benefit in the sunitinib arm [14]. A subsequent analysis from the Checkmate-025 study of patients with mRCC treated in the second-line or beyond and randomized to nivolumab or everolimus showed that there was an enrichment of clinical benefit in the PBRM1 mutant group in nivolumab-treated patients, though this trial did not include a VEGF-targeted therapy. The effect of PBRM1 mutations on response and survival in this study was modest, with median PFS 5.6 vs. 2.9 months (HR, 0.67; 95% CI, 0.47–0.96; p = 0.03) and median OS 27.9 vs. 20.9 months (HR, 0.65; 95% CI, 0.44–0.96; p = 0.03) [15].
Finally, a large retrospective analysis (n = 2936) explored the interaction between PBRM1 mutations and immunotherapy across cancer types and failed to show a statistically significant association with OS (HR 0.9, p = 0.7). Interestingly, this trial included 189 patients with mRCC treated with ICB and this subgroup did demonstrate an association with OS (HR 1.24, p = 0.47). It was previously hypothesized in the initial discovery study by Miao et al. that PBRM1 mutations increased interferon-gamma (IFNγ) gene expression and thereby modulated the immune response. However, this analysis explored the impact of IFNγ signaling in both the cohort studied by McDermott et al. and a cohort from the previously mentioned COMPARZ trial and showed unchanged or decreased IFNγ signaling in PBRM1 mutants compared to the wild-type, which conflicted with the hypothesized mechanism of action [16]. Due to the conflicting nature of these results, doubt has been cast on the potential use of PBRM1 as a biomarker for ICB [17].
Although much focus is on finding mutations associated with a response, it is also useful to examine the opposite phenomenon and identify mutations that are associated with resistance to immunotherapy. This would help route patients to therapies more likely to be beneficial and avoid unnecessary toxicity. For example, in non-small cell lung cancer mutations in STK11 have been identified as predictors of poor responses to ICBs [18]. STK11 is not a useful biomarker for RCC since it is very rarely found in RCC on the order of 0.2% of patients based on data from cBioPortal [19]. A retrospective study of patients with mRCC (n = 75), the majority with clear cell histology (~80%), who received comprehensive genomic profiling (whole-exome and RNA sequencing) as part of routine care, including both immunotherapy and targeted therapy, attempted to identify genomic and transcriptomic correlates of clinical benefit. The authors found that mutations in the TERT promoter were specifically associated with a lack of benefit from ICB. In this subgroup of TERT promoter mutated tumors the authors also found enrichment of transcription factor targets of MYC and KATA2, and kinase targets of CDK4, ATM, and MAPK14 [20].
Similar to approaches in VEGF TKI treated patients, researchers have investigated potential tumor genomic signatures that might serve as predictive biomarkers for ICB. In an exploratory analysis of the IMmotion150 study, the authors used gene signatures previously defined and representing angiogenesis, immune response (T-effector/IFNγ), and myeloid inflammatory gene expression to perform a subgroup analysis and investigate associations with response. They found that tumors with high expression of a T-effector gene signature (TeffHigh) were positively associated with the expression of PD-L1 and CD8 T-cell infiltration. They also showed that within this group there was increased expression of the myeloid inflammation genes. The TeffHigh gene signature was also associated with improved ORR and PFS when compared to the TeffLow group within the atezolizumab/bevacizumab arm. They also showed that TeffHigh was associated with improved PFS when compared across groups to the sunitinib arm. High myeloid inflammation gene signature expression (MyeloidHigh), which had previously been shown to be associated with suppressed T-cell responses, was shown to be associated with worse PFS in both the atezolizumab monotherapy and atezolizumab/bevacizumab arms [14].
A separate group utilized machine learning techniques to build upon the prior IMmotion150 gene signatures to define a specific 66-gene signature created for mRCC using RNA sequencing data from The Cancer Genome Atlas (TCGA) dataset in cBioPortal. They identified that the genes in the IMmotion150 gene signature were selected by analysis of the literature and citations which defined the three biological axes explored in the study and not based on an empirical analysis of the data, which they considered to be a limitation of the previous approach. To develop their signature, they first leveraged the gene signatures defined by the IMmotion150 study to perform unsupervised clustering to categorize patients into three groups and confirmed they separated into the same three categories; angiogenesis, T-effector and myeloid inflammation. They then utilized a separate featured selection machine learning technique to analyze the global gene expression profile of the sub-classified patients and selected the top 500 ranked and subsequently refined them using several different techniques to investigate the underlying biology and came up with their 66-gene signature. Using training and validation cohorts, they were able to show that this signature performed better with regards to association with OS and DFS than the original IMmotion150 signature. However, interpretation of this signature thus far is limited since annotation of treatments record and outcome are not available in the TCGA data and survival data was calculated prior to the approval of ICBs. The signature does, however, hold promise to be tested in cohorts who did receive ICB to test what they hypothesize as an improvement in the clustering of patients into unique groups defined by tumor biology [21].
In an analysis of the results of KEYNOTE-427 (pembrolizumab monotherapy) 11 separate gene signatures were analyzed for associations with response. They identified one signature, a T-cell inflamed gene expression profile (GEP), which stood out demonstrating a strong association with ORR to pembrolizumab. The same T-cell inflamed GEP signature, however, was not associated with longer PFS or OS in the same study and thus remains hypothesis generating [22] (Table 1).
Table 1. Summary of gene expression signatures.
| Gene Signature | Dataset | Genes | Key Findings | |
|---|---|---|---|---|
| IMmotion150 Signature [14] | Sample size: 263 patients Study Type: Randomized phase 2 study of atezolizumab alone or combined with bevacizumab (anti-VEGF) versus sunitinib |
Angiogenesis (Angio) |
|
|
|
|
|||
| Myeloid Inflammation | ||||
|
|
|||
| T-effector (Teff) | ||||
|
|
|||
| 66 Gene Signature [21] | Sample Size: Training cohort 469 patients Validation cohort 64 patients Study Type: Retrospective analysis of ccRCC patients from The Cancer Genome Atlas (TCGA) |
Angiogenesis |
|
|
|
|
|||
| T-effector | ||||
|
|
|||
| Ca2+-flux | ||||
|
|
|||
| Invasion | ||||
|
|
|||
| T-cell Inflamed GEP [22] | Sample Size: 78 patients Study Type: Open-label, single-arm phase 2 study of first-line pembrolizumab |
T-cell Inflamed |
|
|
|
|
|||
Although less common in some other tumor types, RCC can harbor alterations in DNA damage repair (DDR) pathways, including defects in DNA mismatch repair (dMMR). Loss of function of certain genes related to dMMR defects can lead to lead to high levels of microsatellite instability (MSI), which has been established as a biomarker for response to immunotherapy irrespective of tumor type [23]. MSI-Hi tumors are not a common finding in RCC and are estimated to be present in only 1–2% of cases [24]. As a result, MSI is not a practical biomarker in a broad sense for ICB in RCC since many non-MSI RCC tumors respond to immunotherapy.
Looking more broadly, mutations in genes involved in the various DDR pathways, which do not necessarily result in MSI, are relatively prevalent in RCC. In one cohort published by Ged et al., about 19% of patients (43/229) with mRCC harbor DDR mutations, with CHEK2 and ATM being the most frequently mutated. In this cohort, they were able to demonstrate a correlation between DDR mutation status and superior OS (HR 0.41; 95% CI: 0.14–1.14; p = 0.09) in patients treated with ICB [25]. This finding was also reported in a smaller cohort (n = 34) by Labriola et al. who showed that patients with DDR mutant tumors had improved disease control (defined as CR, PR, or SD) with ICB [26].
Another measure of disruption of genomic integrity is tumor mutational burden (TMB). TMB is defined by the total number of non-synonymous alterations (single-nucleotide variants or insertions/deletions) and is typically calculated from next-generation sequencing (NGS) data of either the whole exome or large targeted panels. A high TMB is thought to be integral in promoting increases in the expression of tumor neoantigens which promote T-cell mediated immune responses against tumors [27][28]. TMB, similarly to MSI, has been investigated independent of tumor histology and has been shown to enrich response to ICB [29][30]. This also led to an FDA approval on 16 June 2020 of pembrolizumab for all TMB-high tumors (defined as >10 mutations per megabase) regardless of histology.
However, this approval has been met with controversy because of concerns that cutoffs for TMB and its performance as a biomarker may differ between tumor types. This skepticism is supported in RCC based on some of the available data. For example, in the study discussed above by Labriola et al., which focused solely on RCC, there was no observed association between TMB and disease control in patients treated with ICB [26]. This was also seen in a separate and larger cohort of 592 patients treated with nivolumab (pooled analysis of checkmate 009, 010, and 025) showed no association with response to PD-1 blockade. Paradoxically, it also has been shown that high-TMB is actually associated with inhibition of immune cell infiltrates in RCC tumors, which supports and possibly explains these unexpected clinical observations on a cellular level [31].
Another interesting observation that may help explain why RCC is such an immunogenic tumor but has a characteristically low TMB is the distribution of mutations that comprise its TMB [32][33]. TMB high tumors traditionally have a predominance of many single nucleotide variants (SNVs) making up the majority of mutations, while RCC on the other hand has a uniquely high proportion of insertions and deletions (indels) relative to other tumors. This phenomenon was identified as part of an analysis of the Cancer Genome Atlas study of 5777 solid tumors which identified RCC tumors as having more than double the median proportion of indels to SNVs. The authors then hypothesized that indels are more efficient in the formation of immunogenic peptides serving as neoantigens and using in silico prediction models they were able to show an enrichment of high-affinity neoantigens from indels that were three times that of SNV [34]. This suggests that RCC may be a case of quality over quantity in regards to immunogenic mutations.
Another approach to improving the performance of TMB as a biomarker is incorporating HLA correction. HLA correction is a computational method by which the incorporation of loss of heterozygosity of HLA alleles is thought to improve upon TMB by predicting the proportion of functional neoantigens present. This has been studied in non-small cell lung cancer and shown to identify and reclassify tumors previously characterized as TMB-high and, in doing so, improve the association with the response to ICB, but has yet to be studied in RCC [35].
In the search for biomarkers predictive of response to ICB, the investigation has necessarily expanded beyond clinical- and tumor-dependent factors, such as performance status or genomics, and additionally focused on host-dependent components of the immune system. In order to study the cellular components of the immune system, including T-cells, neutrophils, NK cells, and antigen-presenting cells, a variety of techniques have been approached, ranging from simple analytes (like a complete blood count with differential) to more complex methods like flow cytometry and advanced staining techniques, like multiplex IHC.

