 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Eduardo Perez-Campos | + 1631 word(s) | 1631 | 2021-10-26 08:32:34 | | | |
| 2 | Lindsay Dong | + 7 word(s) | 1638 | 2021-11-22 03:26:59 | | |
Video Upload Options
During COVID-19 infection, SARS-Cov-2 interacts with Angiotensin-Converting Enzyme 2 (ACE2), NRP1, endothelial cells, platelets, neutrophil extracellular traps (NETs), thrombin, extracellular DNA (eDNA), and histones, inducing heterogeneous clinical manifestations characterized by endothelial damage, microthrombosis, and inflammation.
1. Introduction
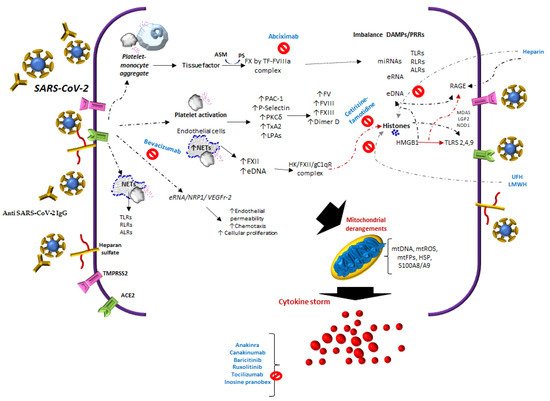
1.1. Factors of the Contact System
In COVID-19, specifically, the expression of FXIIa increases in lung tissue. In addition, this factor is colocalized with NETs in the lungs, indicating that the accumulation of NETs leads to greater activation of FXII due to a defect in the clearance of NETs by DNases, contributing to procoagulant activity [15]. This is also related to the activity of FXII in the blood coagulation system and increases in DNA and H4 histones [16]. Histones contribute to microvascular thrombosis and competitively inhibit plasmin to delay fibrinolysis [17].
1.2. Tissue Factor
In COVID-19, increased circulating extracellular vesicle TF activity has been reported, which correlates with the markers of thrombosis such as D-dimer [18]. It is important to point out that TF expression may be inhibited by platelet P-selectin (CD62P) neutralization or integrin αIIb/β3 blocking, as with abciximab [19].
1.3. Neutrophil Extracellular Traps and Molecule Release
In COVID-19, the platelet/NETs/TF/thrombin axis is enhanced by complement activation [20]. Not only do neutrophils release eDNA, but also macrophages, eosinophils, and mast cells. These appear at different stages of thrombosis [21][22], and similarly in tumor cells [23].
In COVID-19 patients, platelet activation products, such as TXB2 and proteins from platelet α-granules PF4/CXCL4 and PDGF, are also released and found in tracheal aspirates [19].
Of the molecules with NETs that are released, it has been suggested that free DNA may be the cause of a more severe pathology in COVID-19 [24]. Moreover, the manifestations of severity could be related not only to eDNA, but also to other alarmins, such as extracellular heat-shock proteins and HMGB1, mentioned above, in addition to diverse self-nucleic acids, including nuclear DNA, ribosomal RNA, extracellular RNA (eRNA), micro-RNAs, and histones [25], Figure 1.
In COVID-19, eDNA from NETs and histones could also explain thrombosis in severe forms [26]. The half-life of eDNA is around 4–30 min [27]. Its clearance is regulated by different factors, such as 1) Serine proteases, e.g., Factor VII activating protease; cysteine proteases, e.g., caspase-activated DNAse [28]; DNASE1; and deoxyribonuclease 1-like 3 (DNASE1L3) [29].
1.4. Platelets-SARS-CoV-2/Angiotensin-Converting Enzyme 2
Thrombocytopenia in COVID-19 is an indicator of poor prognosis, particularly when it decreases in the first 7 days after admission to the hospital. Thrombocytopenia is an independent risk factor associated with in-hospital mortality. Liu et al. [31] found that an increase of about 50 × 109 /L over the whole range of platelet, decreases mortality. Adding support to the means of platelet activation, several studies show that platelets are activated in COVID-19 patients [32], i.e., there is an increase in young immature platelets named reticulated platelets (RPs). These are associated with high platelet turnover and arterial thrombotic events. In COVID-19, PRs or the immature platelet fraction (IPF) are similar to patients with acute myocardial infarction [33].
1.5. Heparin-Induced Thrombocytopenia
2. Cytokine Storm Syndrome
3. Influence of Heparanase, Heparin and Heparinoids in Complications from COVID-19
References
- Levi, M.; Thachil, J. Coronavirus Disease 2019 Coagulopathy: Disseminated Intravascular Coagulation and Thrombotic Microangiopathy-Either, Neither, or Both. Semin. Thromb. Hemost. 2020, 46, 781–784.
- Gavriilaki, E.; Brodsky, R.A. Severe COVID-19 infection and thrombotic microangiopathy: Success does not come easily. Br. J. Haematol. 2020, 189, e227–e230.
- Falter, T.; Rossmann, H.; Menge, P.; Goetje, J.; Groenwoldt, S.; Weinmann, A.; Sivanathan, V.; Schulz, A.; Lemmermann, N.A.W.; Danckwardt, S.; et al. No Evidence for Classic Thrombotic Microangiopathy in COVID-19. J. Clin. Med. 2021, 10, 671.
- Venter, C.; Bezuidenhout, J.A.; Laubscher, G.J.; Lourens, P.J.; Steenkamp, J.; Kell, D.B.; Pretorius, E. Erythrocyte, Platelet, Serum Ferritin, and P-Selectin Pathophysiology Implicated in Severe Hypercoagulation and Vascular Complications in COVID-19. Int. J. Mol. Sci. 2020, 21, 8234.
- Zhang, S.; Liu, Y.; Wang, X.; Yang, L.; Li, H.; Wang, Y.; Liu, M.; Zhao, X.; Xie, Y.; Yang, Y.; et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J. Hematol. Oncol. 2020, 13, 120.
- Wright, F.L.; Vogler, T.O.; Moore, E.E.; Moore, H.B.; Wohlauer, M.V.; Urban, S.; Nydam, T.L.; Moore, P.K.; McIntyre, R.C., Jr. Fibrinolysis Shutdown Correlation with Thromboembolic Events in Severe COVID-19 Infection. J. Am. Coll. Surg. 2020, 231, 193–203.
- Bachler, M.; Bösch, J.; Stürzel, D.P.; Hell, T.; Giebl, A.; Ströhle, M.; Klein, S.J.; Schäfer, V.; Lehner, G.F.; Joannidis, M.; et al. Impaired fibrinolysis in critically ill COVID-19 patients. Br. J. Anaesth. 2021, 126, 590–598.
- Carsana, L.; Sonzogni, A.; Nasr, A.; Rossi, R.S.; Pellegrinelli, A.; Zerbi, P.; Rech, R.; Colombo, R.; Antinori, S.; Corbellino, M.; et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: A two-centre descriptive study. Lancet Infect. Dis. 2020, 20, 1135–1140.
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1038.
- Asakura, H.; Ogawa, H. COVID-19-associated coagulopathy and disseminated intravascular coagulation. Int. J. Hematol. 2021, 113, 45–57.
- Miesbach, W.; Makris, M. COVID-19: Coagulopathy, Risk of Thrombosis, and the Rationale for Anticoagulation. Clin. Appl. Thromb. Hemost. 2020, 26, 1076029620938149.
- Beristain-Covarrubias, N.; Perez-Toledo, M.; Thomas, M.R.; Henderson, I.R.; Watson, S.P.; Cunningham, A.F. Understanding Infection-Induced Thrombosis: Lessons Learned From Animal Models. Front. Immunol. 2019, 10, 2569.
- Jayarangaiah, A.; Kariyanna, P.T.; Chen, X.; Jayarangaiah, A.; Kumar, A. COVID-19-Associated Coagulopathy: An Exacerbated Immunothrombosis Response. Clin. Appl. Thromb. Hemost. 2020, 26, 1076029620943293.
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Investig. 2020, 130, 6151–6157.
- Englert, H.; Rangaswamy, C.; Deppermann, C.; Sperhake, J.P.; Krisp, C.; Schreier, D.; Gordon, E.; Konrath, S.; Haddad, M.; Pula, G.; et al. Defective NET clearance contributes to sustained FXII activation in COVID-19-associated pulmonary thrombo-inflammation. EBioMedicine 2021, 67, 103382.
- Reyes-García, A.M.L.; Aroca, A.; Arroyo, A.B.; García-Barbera, N.; Vicente, V.; González-Conejero, R.; Martínez, C. Neutrophil extracellular trap components increase the expression of coagulation factors. Biomed. Rep. 2019, 10, 195–201.
- Locke, M.; Longstaff, C. Extracellular Histones Inhibit Fibrinolysis through Noncovalent and Covalent Interactions with Fibrin. Thromb. Haemost. 2021, 121, 464–476.
- Rosell, A.; Havervall, S.; von Meijenfeldt, F.; Hisada, Y.; Aguilera, K.; Grover, S.P.; Lisman, T.; Mackman, N.; Thålin, C. Patients With COVID-19 Have Elevated Levels of Circulating Extracellular Vesicle Tissue Factor Activity That Is Associated With Severity and Mortality-Brief Report. Arter. Thromb. Vasc. Biol. 2021, 41, 878–882.
- Hottz, E.D.; Azevedo-Quintanilha, I.G.; Palhinha, L.; Teixeira, L.; Barreto, E.A.; Pão, C.R.R.; Righy, C.; Franco, S.; Souza, T.M.L.; Kurtz, P.; et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood 2020, 136, 1330–1341.
- Veras, F.P.; Pontelli, M.C.; Silva, C.M.; Toller-Kawahisa, J.E.; de Lima, M.; Nascimento, D.C.; Schneider, A.H.; Caetité, D.; Tavares, L.A.; Paiva, I.M.; et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J. Exp. Med. 2020, 217, e20201129.
- Daniel, C.; Leppkes, M.; Muñoz, L.E.; Schley, G.; Schett, G.; Herrmann, M. Extracellular DNA traps in inflammation, injury and healing. Nat. Rev. Nephrol. 2019, 15, 559–575.
- Pertiwi, K.R.; de Boer, O.J.; Mackaaij, C.; Pabittei, D.R.; de Winter, R.J.; Li, X.; van der Wal, A.C. Extracellular traps derived from macrophages, mast cells, eosinophils and neutrophils are generated in a time-dependent manner during atherothrombosis. J. Pathol. 2019, 247, 505–512.
- Tan, S.; Davey, C.A. Nucleosome structural studies. Curr. Opin. Struct. Biol. 2011, 21, 128–136.
- Liu, B. Free DNA, a reason for severe COVID-19 infection? Med. Hypotheses 2020, 142, 109812.
- Preissner, K.T.; Fischer, S.; Deindl, E. Extracellular RNA as a Versatile DAMP and Alarm Signal That Influences Leukocyte Recruitment in Inflammation and Infection. Front. Cell Dev. Biol. 2020, 8, 619221.
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885.
- Lo, Y.M.; Zhang, J.; Leung, T.N.; Lau, T.K.; Chang, A.M.; Hjelm, N.M. Rapid clearance of fetal DNA from maternal plasma. Am. J. Hum. Genet. 1999, 64, 218–224.
- Larsen, B.D.; Sørensen, C.S. The caspase-activated DNase: Apoptosis and beyond. FEBS J. 2017, 284, 1160–1170.
- Jiménez-Alcázar, M.; Rangaswamy, C.; Panda, R.; Bitterling, J.; Simsek, Y.J.; Long, A.T.; Bilyy, R.; Krenn, V.; Renné, C.; Renné, T.; et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science 2017, 358, 1202–1206.
- 56. Okur, H.K.; Yalcin, K.; Tastan, C.; Demir, S.; Yurtsever, B.; Karakus, G.S.; Kancagi, D.D.; Abanuz, S.; Seyis, U.; Zengin, R.; et al. Preliminary report of in vitro and in vivo effectiveness of dornase alfa on SARS-CoV-2 infection. New Microbes New Infect. 2020, 37, 100756.
- Liu, Y.; Sun, W.; Guo, Y.; Chen, L.; Zhang, L.; Zhao, S.; Long, D.; Yu, L. Association between platelet parameters and mortality in coronavirus disease 2019: Retrospective cohort study. Platelets 2020, 31, 490–496.
- Campbell, R.A.; Boilard, E.; Rondina, M.T. Is there a role for the ACE2 receptor in SARS-CoV-2 interactions with platelets? J. Thromb. Haemost. 2021, 19, 46–50.
- Cohen, A.; Harari, E.; Cipok, M.; Laish-Farkash, A.; Bryk, G.; Yahud, E.; Sela, Y.; Lador, N.K.; Mann, T.; Mayo, A.; et al. Immature platelets in patients hospitalized with Covid-19. J. Thromb. Thrombolysis 2021, 51, 608–616.
- Sartori, M.; Cosmi, B. Heparin-induced thrombocytopenia and COVID-19. Hematol. Rep. 2021, 13, 8857.
- Lingamaneni, P.; Gonakoti, S.; Moturi, K.; Vohra, I.; Zia, M. Heparin-Induced Thrombocytopenia in COVID-19. J. Investig. Med. High Impact Case Rep. 2020, 8, 2324709620944091.
- Daviet, F.; Guervilly, C.; Baldesi, O.; Bernard-Guervilly, F.; Pilarczyk, E.; Genin, A.; Lefebvre, L.; Forel, J.M.; Papazian, L.; Camoin-Jau, L. Heparin-Induced Thrombocytopenia in Severe COVID-19. Circulation 2020, 142, 1875–1877.
- Warkentin, T.E.; Kaatz, S. COVID-19 versus HIT hypercoagulability. Thromb. Res. 2020, 196, 38–51.
- Uaprasert, N.; Tangcheewinsirikul, N.; Rojnuckarin, P.; Patell, R.; Zwicker, J.I.; Chiasakul, T. Heparin-induced Thrombocytopenia in Patients with Coronavirus Disease 2019: Systematic Review and Meta-analysis. Blood Adv. 2021.
- Henderson, L.A.; Canna, S.W.; Schulert, G.S.; Volpi, S.; Lee, P.Y.; Kernan, K.F.; Caricchio, R.; Mahmud, S.; Hazen, M.M.; Halyabar, O.; et al. On the Alert for Cytokine Storm: Immunopathology in COVID-19. Arthritis Rheumatol. 2020, 72, 1059–1063.
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.
- Vanderheiden, A.; Ralfs, P.; Chirkova, T.; Upadhyay, A.A.; Zimmerman, M.G.; Bedoya, S.; Aoued, H.; Tharp, G.M.; Pellegrini, K.L.; Manfredi, C.; et al. Type I and Type III Interferons Restrict SARS-CoV-2 Infection of Human Airway Epithelial Cultures. J. Virol. 2020, 94, e0098520.
- Zheng, Y.; Zhuang, M.W.; Han, L.; Zhang, J.; Nan, M.L.; Zhan, P.; Kang, D.; Liu, X.; Gao, C.; Wang, P.H. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) membrane (M) protein inhibits type I and III interferon production by targeting RIG-I/MDA-5 signaling. Signal. Transduct. Target. Ther. 2020, 5, 299.
- Burgos-Blasco, B.; Güemes-Villahoz, N.; Santiago, J.L.; Fernandez-Vigo, J.I.; Espino-Paisán, L.; Sarriá, B.; García-Feijoo, J.; Martinez-de-la-Casa, J.M. Hypercytokinemia in COVID-19: Tear cytokine profile in hospitalized COVID-19 patients. Exp. Eye Res. 2020, 200, 108253.
- Sang, Y.; Miller, L.C.; Blecha, F. Macrophage Polarization in Virus-Host Interactions. J. Clin. Cell Immunol. 2015, 6, 311.
- Mahajan, S.; Decker, C.E.; Yang, Z.; Veis, D.; Mellins, E.D.; Faccio, R. Plcγ2/Tmem178 dependent pathway in myeloid cells modulates the pathogenesis of cytokine storm syndrome. J. Autoimmun. 2019, 100, 62–74.
- Holter, J.C.; Pischke, S.E.; de Boer, E.; Lind, A.; Jenum, S.; Holten, A.R.; Tonby, K.; Barratt-Due, A.; Sokolova, M.; Schjalm, C.; et al. Systemic complement activation is associated with respiratory failure in COVID-19 hospitalized patients. Proc. Natl. Acad. Sci. USA 2020, 117, 25018–25025.
- Soy, M.; Keser, G.; Atagündüz, P.; Tabak, F.; Atagündüz, I.; Kayhan, S. Cytokine storm in COVID-19: Pathogenesis and overview of anti-inflammatory agents used in treatment. Clin. Rheumatol. 2020, 39, 2085–2094.
- Hussman, J.P. Severe Clinical Worsening in COVID-19 and Potential Mechanisms of Immune-Enhanced Disease. Front. Med. 2021, 8, 637642.
- Cardone, M.; Yano, M.; Rosenberg, A.S.; Puig, M. Lessons Learned to Date on COVID-19 Hyperinflammatory Syndrome: Considerations for Interventions to Mitigate SARS-CoV-2 Viral Infection and Detrimental Hyperinflammation. Front. Immunol. 2020, 11, 1131.
- Kaur, S.; Bansal, R.; Kollimuttathuillam, S.; Gowda, A.M.; Singh, B.; Mehta, D.; Maroules, M. The looming storm: Blood and cytokines in COVID-19. Blood Rev. 2020, 46, 100743.
- Quartuccio, L.; Sonaglia, A.; McGonagle, D.; Fabris, M.; Peghin, M.; Pecori, D.; De Monte, A.; Bove, T.; Curcio, F.; Bassi, F.; et al. Profiling COVID-19 pneumonia progressing into the cytokine storm syndrome: Results from a single Italian Centre study on tocilizumab versus standard of care. J. Clin. Virol. 2020, 129, 104444.
- Didangelos, A. COVID-19 Hyperinflammation: What about Neutrophils? mSphere 2020, 5, e00367.
- Mokhtari, T.; Hassani, F.; Ghaffari, N.; Ebrahimi, B.; Yarahmadi, A.; Hassanzadeh, G. COVID-19 and multiorgan failure: A narrative review on potential mechanisms. J. Mol. Histol. 2020, 51, 613–628.
- Kim, J.S.; Lee, J.Y.; Yang, J.W.; Lee, K.H.; Effenberger, M.; Szpirt, W.; Kronbichler, A.; Shin, J.I. Immunopathogenesis and treatment of cytokine storm in COVID-19. Theranostics 2021, 11, 316–329.
- Monteagudo, L.A.; Boothby, A.; Gertner, E. Continuous Intravenous Anakinra Infusion to Calm the Cytokine Storm in Macrophage Activation Syndrome. ACR Open Rheumatol. 2020, 2, 276–282.
- Ucciferri, C.; Auricchio, A.; Di Nicola, M.; Potere, N.; Abbate, A.; Cipollone, F.; Vecchiet, J.; Falasca, K. Canakinumab in a subgroup of patients with COVID-19. Lancet Rheumatol. 2020, 2, e457–e458.
- Kalil, A.C.; Patterson, T.F.; Mehta, A.K.; Tomashek, K.M.; Wolfe, C.R.; Ghazaryan, V.; Marconi, V.C.; Ruiz-Palacios, G.M.; Hsieh, L.; Kline, S.; et al. Baricitinib plus Remdesivir for Hospitalized Adults with Covid-19. N. Engl. J. Med. 2021, 384, 795–807.
- Kaplanski, G.; Bontemps, D.; Esnault, P.; Blasco, V.; Carvelli, J.; Delarbre, D.; Cauchois, R.; Forel, J.M.; Papazian, L. Combined Anakinra and Ruxolitinib treatment to rescue extremely ill COVID-19 patients: A pilot study. Autoimmun. Rev. 2021, 20, 102726.
- Salama, C.; Han, J.; Yau, L.; Reiss, W.G.; Kramer, B.; Neidhart, J.D.; Criner, G.J.; Kaplan-Lewis, E.; Baden, R.; Pandit, L.; et al. Tocilizumab in Patients Hospitalized with Covid-19 Pneumonia. N. Engl. J. Med. 2021, 384, 20–30.
- Leaf, D.E.; Gupta, S.; Wang, W. Tocilizumab in Covid-19. N. Engl. J. Med. 2021, 384, 86–87.
- Emadi, A.; Chua, J.V.; Talwani, R.; Bentzen, S.M.; Baddley, J. Safety and Efficacy of Imatinib for Hospitalized Adults with COVID-19: A structured summary of a study protocol for a randomised controlled trial. Trials 2020, 21, 897.
- Petrova, M.; Jelev, D.; Ivanova, A.; Krastev, Z. Isoprinosine affects serum cytokine levels in healthy adults. J. Interf. Cytokine Res. 2010, 30, 223–228.
- Lasek, W.; Janyst, M.; Wolny, R.; Zapała, Ł.; Bocian, K.; Drela, N. Immunomodulatory effects of inosine pranobex on cytokine production by human lymphocytes. Acta Pharm. 2015, 65, 171–180.
- Beran, J.; Špajdel, M.; Katzerová, V.; Holoušová, A.; Malyš, J.; Finger-Rousková, J.; Slíva, J. Inosine Pranobex Significantly Decreased the Case-Fatality Rate among PCR Positive Elderly with SARS-CoV-2 at Three Nursing Homes in the Czech Republic. Pathogens 2020, 9, 1055.
- Wu, L.; Davies, G.J. An Overview of the Structure, Mechanism and Specificity of Human Heparanase. Adv. Exp. Med. Biol. 2020, 1221, 139–167.
- Nadir, Y.; Brenner, B.; Fux, L.; Shafat, I.; Attias, J.; Vlodavsky, I. Heparanase enhances the generation of activated factor X in the presence of tissue factor and activated factor VII. Haematologica 2010, 95, 1927–1934.
- Cui, H.; Tan, Y.X.; Österholm, C.; Zhang, X.; Hedin, U.; Vlodavsky, I.; Li, J.P. Heparanase expression upregulates platelet adhesion activity and thrombogenicity. Oncotarget 2016, 7, 39486–39496.
- Buijsers, B.; Yanginlar, C.; de Nooijer, A.; Grondman, I.; Maciej-Hulme, M.L.; Jonkman, I.; Janssen, N.A.F.; Rother, N.; de Graaf, M.; Pickkers, P.; et al. Increased Plasma Heparanase Activity in COVID-19 Patients. Front. Immunol. 2020, 11, 575047.
- Paar, V.; Wernly, B.; Zhou, Z.; Motloch, L.J.; Hoppe, U.C.; Egle, A.; Lichtenauer, M. Anti-coagulation for COVID-19 treatment: Both anti-thrombotic and anti-inflammatory? J. Thromb. Thrombolysis 2021, 51, 226–231.
- Buijsers, B.; Yanginlar, C.; Maciej-Hulme, M.L.; de Mast, Q.; van der Vlag, J. Beneficial non-anticoagulant mechanisms underlying heparin treatment of COVID-19 patients. EBioMedicine 2020, 59, 102969.
- Garcia, D.A.; Baglin, T.P.; Weitz, J.I.; Samama, M.M. Parenteral anticoagulants: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2013, 141 (Suppl. 2), e24S–e43S, Erratum in 2013, 144, 721.
- Longstaff, C.; Hogwood, J.; Gray, E.; Komorowicz, E.; Varjú, I.; Varga, Z.; Kolev, K. Neutralisation of the anti-coagulant effects of heparin by histones in blood plasma and purified systems. Thromb. Haemost. 2016, 115, 591–599.
- Kilgore, K.S.; Tanhehco, E.J.; Naylor, K.B.; Lucchesi, B.R. Ex vivo reversal of heparin-mediated cardioprotection by heparinase after ischemia and reperfusion. J. Pharm. Exp. Ther. 1999, 290, 1041–1047.
- Hippensteel, J.A.; LaRiviere, W.B.; Colbert, J.F.; Langouët-Astrié, C.J.; Schmidt, E.P. Heparin as a therapy for COVID-19: Current evidence and future possibilities. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 319, L211–L217.
- Stattin, K.; Lipcsey, M.; Andersson, H.; Pontén, E.; Bülow Anderberg, S.; Gradin, A.; Larsson, A.; Lubenow, N.; von Seth, M.; Rubertsson, S.; et al. Inadequate prophylactic effect of low-molecular weight heparin in critically ill COVID-19 patients. J. Crit. Care 2020, 60, 249–252.
- Capell, W.H.; Barnathan, E.S.; Piazza, G.; Spyropoulos, A.C.; Hsia, J.; Bull, S.; Lipardi, C.; Sugarmann, C.; Suh, E.; Rao, J.P.; et al. Rationale and design for the study of rivaroxaban to reduce thrombotic events, hospitalization and death in outpatients with COVID-19: The PREVENT-HD study. Am. Heart J. 2021, 235, 12–23.
- Akel, T.; Qaqa, F.; Abuarqoub, A.; Shamoon, F. Pulmonary embolism: A complication of COVID 19 infection. Thromb. Res. 2020, 193, 79–82.
- Gotts, J.E.; Matthay, M.A. Sepsis: Pathophysiology and clinical management. BMJ 2016, 353, i1585.
- Taeb, A.M.; Hooper, M.H.; Marik, P.E. Sepsis: Current Definition, Pathophysiology, Diagnosis, and Management. Nutr. Clin. Pract. 2017, 32, 296–308.
- Clausen, T.M.; Sandoval, D.R.; Spliid, C.B.; Pihl, J.; Perrett, H.R.; Painter, C.D.; Narayanan, A.; Majowicz, S.A.; Kwong, E.M.; McVicar, R.N.; et al. SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell 2020, 183, 1043–1057.
- Mycroft-West, C.J.; Su, D.; Pagani, I.; Rudd, T.R.; Elli, S.; Gandhi, N.S.; Guimond, S.E.; Miller, G.J.; Meneghetti, M.C.Z.; Nader, H.B.; et al. Heparin Inhibits Cellular Invasion by SARS-CoV-2: Structural Dependence of the Interaction of the Spike S1 Receptor-Binding Domain with Heparin. Thromb. Haemost. 2020, 120, 1700–1715.
- Nandy, A.; Mondal, T.; Sarkar, M.; Nag, S.S.; Chel, S.; Ivan, D.M.; Hazra, A.; Mondal, R. Multiorgan dysfunction syndrome in sepsis: Is macrophage activation syndrome secondary to infection? Eur. J. Rheumatol. 2020, 8, 89–92.
- Marsman, G.; von Richthofen, H.; Bulder, I.; Lupu, F.; Hazelzet, J.; Luken, B.M.; Zeerleder, S. DNA and factor VII-activating protease protect against the cytotoxicity of histones. Blood Adv. 2017, 1, 2491–2502.
- Yamamichi, S.; Fujiwara, Y.; Kikuchi, T.; Nishitani, M.; Matsushita, Y.; Hasumi, K. Extracellular histone induces plasma hyaluronan-binding protein (factor VII activating protease) activation in vivo. Biochem. Biophys. Res. Commun. 2011, 409, 483–488.
- Cosmi, B.; Cini, M.; Legnani, C.; Pancani, C.; Calanni, F.; Coccheri, S. Additive thrombin inhibition by fast moving heparin and dermatan sulfate explains the anticoagulant effect of sulodexide, a natural mixture of glycosaminoglycans. Thromb. Res. 2003, 109, 333–339.
- Siddiqui, F.; Hoppensteadt, D.; Bontekoe, E.; Farooqui, A.; Jeske, W.; Fareed, J. Comparative Anticoagulant and Thrombin Generation Inhibitory Profile of Heparin, Sulodexide and Its Components. Clin. Appl. Thromb. Hemost. 2020, 26, 1076029620954913.
- De Felice, F.; Megiorni, F.; Pietrantoni, I.; Tini, P.; Lessiani, G.; Mastroiacovo, D.; Mattana, P.; Antinozzi, C.; Di Luigi, L.; Delle Monache, S.; et al. Sulodexide counteracts endothelial dysfunction induced by metabolic or non-metabolic stresses through activation of the autophagic program. Eur. Rev. Med. Pharm. Sci. 2019, 23, 2669–2680.
- Borawski, J.; Gozdzikiewicz, J.; Dubowski, M.; Pawlak, K.; Mysliwiec, M. Tissue factor pathway inhibitor release and depletion by sulodexide in humans. Adv. Med. Sci. 2009, 54, 32–36.
- White, D.; MacDonald, S.; Edwards, T.; Bridgeman, C.; Hayman, M.; Sharp, M.; Cox-Morton, S.; Duff, E.; Mahajan, S.; Moore, C.; et al. Evaluation of COVID-19 coagulopathy; laboratory characterization using thrombin generation and nonconventional haemostasis assays. Int. J. Lab. Hematol. 2021, 43, 123–130.
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.E.; Williamson, M.K.; Antón-Plágaro, C.; Shoemark, D.K.; Simón-Gracia, L.; Bauer, M.; Hollandi, R.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 2020, 370, 861–865.
- Liaw, P.C.; Becker, D.L.; Stafford, A.R.; Fredenburgh, J.C.; Weitz, J.I. Molecular basis for the susceptibility of fibrin-bound thrombin to inactivation by heparin cofactor ii in the presence of dermatan sulfate but not heparin. J. Biol. Chem. 2001, 276, 20959–20965.


